U.S. Dept. of Commerce / NOAA / OAR / PMEL / Publications
Sulfur Emissions to the Atmosphere from Natural Sources
T. S. Bates
NOAA/Pacific Marine Environmental Laboratory, Seattle, WA 98115
B. K. Lamb
Laboratory for Atmospheric Research, Washington State University, Pullman, WA 99164
A. Guenther
Cooperative Institute for Research in the Environmental Sciences, University of
Colorado, Boulder, CO 80303
J. Dignon
Lawrence Livermore National Laboratory, Livermore, CA 94550
R. E. Stoiber
Department of Earth Sciences, Dartmouth College, Hanover, NH 03755
Journal of Atmospheric Chemistry, 14, 315-337, 1992
Copyright ©1992 Kluwer Academic Publishers. Further electronic distribution is not
allowed.
2. Estimates of Natural Emissions
2.1. Marine
2.1.1. Oceanic concentrations of volatile sulfur gases. Several
volatile sulfur gases are produced biologically (CH SCH,
CS
SCH,
CS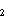 , CHSH,
CHSSCH)
and/or photochemically (HS, OCS)
in the surface waters of the ocean (Andreae,
1986). Of these compounds, dimethyl-sulfide (CHSCH or DMS) is the most abundant and is the only compound which contributes significantly
to the atmospheric burden of NSS sulfate (Cline
and Bates, 1983; Nguyen et al., 1983; Andreae,
1986).
, CHSH,
CHSSCH)
and/or photochemically (HS, OCS)
in the surface waters of the ocean (Andreae,
1986). Of these compounds, dimethyl-sulfide (CHSCH or DMS) is the most abundant and is the only compound which contributes significantly
to the atmospheric burden of NSS sulfate (Cline
and Bates, 1983; Nguyen et al., 1983; Andreae,
1986).
Haas first identified DMS emissions from the marine algae Polysiphonia fastigiata in 1935. The precursor of DMS, dimethyl-sulfoniopropionate (DMSP), was isolated
from this species by Challenger
and Simpson (1948) and the biosynthesis of DMSP from methionine has been
demonstrated by Green
(1962). The occurrence of DMSP in marine phytoplankton was first documented
by Ackman
and coworkers (1966). The DMSP concentration within phytoplankton cells
is extremely species specific (Keller et al., 1989) and has been shown to contribute significantly to the
intracellular osmotic pressure (Reed,
1983; Vairavamurthy et al., 1985), thus serving to maintain the osmotic balance required
for cell growth. DMSP is released from phytoplankton cells both during senescence (Nguyen et al., 1988; Leck et al., 1990) and during zooplankton grazing (Dacey
and Wakeham, 1986; Leck et al., 1990) and is then cleaved enzymatically to yield DMS and
acrylic acid (Cantoni
and Anderson, 1956; Dacey
and Blough, 1987).
DMS was reported first in oceanic waters by Lovelock
and coworkers (1972) and has subsequently been measured throughout the Pacific (Andreae
and Raemdonck, 1983; Cline
and Bates, 1983; Barnard et al., 1984; Andreae,
1985; Bates
and Cline, 1985; Bates et al., 1987), Atlantic (Barnard et al., 1982; Andreae
and Barnard, 1984; Turner
and Liss, 1985; Turner et al., 1988; Iverson et al., 1989), and Southern Oceans (Berresheim,
1987; Berresheim et al., 1990). DMS concentrations vary substantially on a regional
and seasonal basis, and have been recently summarized by Bates
and coworkers (1987) and Cooper
and Matrai (1989). Open ocean surface seawater DMS concentrations generally
range from 0.5 to 5 nmol/L, and are lowest during the winter months in high
latitudes and highest during the summer months at these same latitudes (Bates et al., 1987). On a regional and seasonal basis, the average natural
variability of DMS concentrations in the North Pacific Ocean is approximately
30% (Bates et al., 1987). This includes the analytical uncertainty of approximately
10%. The natural variability in DMS concentrations can be much higher in coastal
regions where concentrations can vary by an order of magnitude within a two-week
period in a given area (Bates
and Cline, 1985; Turner et al., 1989; Leck et al., 1990). The lifetime of DMS in surface waters is on the order
of one day (Kiene
and Bates, 1990) as it is degraded both microbially (Suylen et al., 1986; Taylor
and Kiene, 1989) and photochemically (Brimblecombe
and Shooter, 1986) and lost to the atmosphere via air-sea exchange.
The other reduced sulfur gases in surface ocean waters have been much less
studied. CS is present in surface
open ocean waters at concentrations of approximately 16±8 pmol/L (Kim
and Andreae, 1987; Lovelock,
1974). The sources and sinks of this compound are largely unknown. OCS concentrations
in surface seawater range from 10 to 100 pmol/L (Rasmussen et al., 1982; Ferek
and Andreae, 1983; Turner
and Liss, 1985; Johnson
and Harrison, 1986). The concentrations can vary diurnally by almost an
order of magnitude due to the photochemical production of OCS from organic matter.
The source material may be particulate organic sulfur (Ferek
and Andreae, 1984) which has been measured in surface seawater at concentrations
ranging from 10-100 nmol/L (Matrai,
1989). The hydrolysis of OCS (Elliott et al., 1987) and the decay of sulfur compounds in organic matter (Cutter
and Krahforst, 1988) lead to the formation of the sulfide ion. Although
the sulfide ion is clearly present in surface seawater in concentrations ranging
from 0.1 to 1.1 nmol/L (Cutter
and Krahforst, 1988), it is likely complexed with trace metals (Elliott,
1988; Dyrssen,
1989; Elliott et al., 1989; Elliott
and Rowland, 1990), and hence, not in a volatile form. The other potentially
important volatile species are methanethiol (CHSH)
and dimethyldisulfide (CHSSCH).
Methanethiol is produced in anoxic marine sediments (Kiene
and Visscher, 1987; Sorensen,
1988), in decomposing algal mats (Zinder et al., 1977), and in coastal waters (Leck
and Rodhe, 1990), but as yet has not been quantified in open ocean surface
seawater. Our experience has shown that methanethiol and dimethyldisulfide can
be produced as an artifact of sampling/analysis if plankton cells undergo anaerobic
decomposition (Bates, unpublished data).
2.1.2. Oceanic emissions of volatile sulfur gases. The amount
of oceanic sulfur released to the atmosphere was a major uncertainty in early
atmospheric sulfur budgets. Model estimates ranged from 9 Tmol/a (Eriksson,
1963) to 1 Tmol/a (Granat et al., 1976) and were based solely on the amount of additional
sulfur needed to balance the global budget. This wide range of emissions made
it extremely difficult to assess the significance of anthropogenic sulfur emissions
which were estimated during the early 1970's at approximately 2 Tmol/a (Varhelyi,
1985 and references therein). Although there are still no reliable methods
of directly measuring the flux of volatile oceanic sulfur to the atmosphere (Andreae,
1985), the flux can now be calculated from measured seawater and atmospheric
sulfur gas concentrations. This eliminates a great deal of the uncertainty from
the early model estimates.
The flux of gases across the air-sea interface is calculated from air-sea exchange
models (Liss,
1973) which predict that the flux is proportional to the product of the
concentration difference across the air-sea interface and a first-order exchange
coefficient (V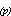 ) such
that:
) such
that:
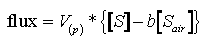 (1)
(1)
where b[S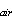 ] is
the equilibrium solubility concentration, (b = Bunsen coefficient, [S]
= atmospheric partial pressure) and [S] is the measured concentration
of the sulfur gas in the surface ocean. For DMS, the dominant volatile sulfur
compound, the concentration calculated assuming equilibrium solubility is quite
small relative to the observed surface water DMS values (Cline
and Bates, 1983; Barnard et al., 1982), hence the flux of DMS = V*[DMS].
The exchange coefficient is a function of both wind speed (Smethie et al., 1985; Liss
and Merlivat, 1986) and the molecular diffusivity of the gas (Holmen
and Liss, 1984; Ledwell,
1984), and has been summarized for DMS by Bates
and coworkers (1987). The root mean square of the uncertainties in these
flux calculations is approximately 66%, based on the standard deviation of the
seasonally averaged concentrations (30%), the range of potential exchange coefficients
(30%), and the calculated molecular diffusivity (50%).
] is
the equilibrium solubility concentration, (b = Bunsen coefficient, [S]
= atmospheric partial pressure) and [S] is the measured concentration
of the sulfur gas in the surface ocean. For DMS, the dominant volatile sulfur
compound, the concentration calculated assuming equilibrium solubility is quite
small relative to the observed surface water DMS values (Cline
and Bates, 1983; Barnard et al., 1982), hence the flux of DMS = V*[DMS].
The exchange coefficient is a function of both wind speed (Smethie et al., 1985; Liss
and Merlivat, 1986) and the molecular diffusivity of the gas (Holmen
and Liss, 1984; Ledwell,
1984), and has been summarized for DMS by Bates
and coworkers (1987). The root mean square of the uncertainties in these
flux calculations is approximately 66%, based on the standard deviation of the
seasonally averaged concentrations (30%), the range of potential exchange coefficients
(30%), and the calculated molecular diffusivity (50%).
The first global ocean-to-atmosphere DMS flux estimates, based on observations
in the Atlantic and Eastern Tropical Pacific, suggested a value near 1 Tmol/a (Andreae
and Raemdonck, 1983; Galloway,
1985; Andreae,
1986). More recent calculations, taking into account the seasonality of
DMS concentrations and a much larger data base, have reduced the calculated
global flux to 0.5 ± 0.33 Tmol/a (Bates et al., 1987). The flux of OCS (Rasmussen et al., 1982; Ferek
and Andreae, 1984; Johnson
and Harrison, 1986) and CS (Kim
and Andreae, 1987) have been estimated at 1-2% of the DMS flux. Presently,
the major uncertainty in calculating the total air-sea exchange of volatile
sulfur is in the flux of HS. Recent
atmospheric measurements by Saltzman
and Cooper (1988) suggest that oceanic HS
may account for approximately 10% of the NSS sulfate in the marine boundary
layer. The implication is that the flux of HS
from the ocean to the atmosphere may be as high as 10% of the DMS flux. Also
in question at this time is the calculated diffusivity of DMS, which has recently
been measured at a level approximately 75% lower than previous estimates (Saltzman et al., 1988; Asher et al., 1990). This would result in an oceanic DMS flux of approximately
0.2-0.3 Tmol/a (Cooper
and Saltzman, 1990).
If your browser cannot view the following table correctly,
click this link for a GIF image of Table 1.
TABLE 1. Oceanic DMS fluxes
|
|
|
|
PACIFIC |
ATLANTIC |
INDIAN |
|
winter |
summer |
winter |
summer |
winter |
summer |
winter |
summer |
|
emiss |
emiss |
flux |
flux |
flux |
flux |
flux |
flux |
| Region |
(µmol S/m˛/d) |
------------------------------(10 mol/d)------------------------------ mol/d)------------------------------ |
|
| 80°–65°N |
0.00 |
2.11 |
0 |
0 |
0 |
19 |
0 |
0 |
| 65°–50°N |
1.40 |
6.57 |
8 |
39 |
8 |
39 |
0 |
0 |
| 50°–35°N |
2.21 |
4.96 |
30 |
68 |
19 |
42 |
0 |
0 |
| 35°–20°N |
2.15 |
5.14 |
44 |
105 |
26 |
63 |
2 |
4 |
| 20°–5°N |
4.72 |
4.45 |
136 |
128 |
51 |
48 |
35 |
33 |
| 5°N–0° |
4.36 |
4.01 |
45 |
41 |
16 |
15 |
14 |
13 |
| 0°–5°S |
4.36 |
4.01 |
42 |
39 |
14 |
13 |
16 |
15 |
| 5°–20°S |
4.72 |
4.45 |
124 |
117 |
42 |
40 |
68 |
64 |
| 20°–35°S |
2.15 |
5.14 |
47 |
113 |
22 |
52 |
29 |
69 |
| 35°–50°S |
2.21 |
4.96 |
40 |
89 |
24 |
55 |
37 |
84 |
| 50°–65°S |
1.40 |
6.57 |
19 |
89 |
12 |
57 |
18 |
83 |
| 65°–80°S |
0.00 |
2.11 |
0 |
11 |
0 |
6 |
0 |
3 |
| N. Hemisphere |
264 |
382 |
120 |
225 |
50 |
49 |
| S. Hemisphere |
271 |
457 |
114 |
222 |
168 |
317 |
|
| Fluxes are the product of the emission rate (Bates et al., 1987) and the ocean area (Levitus,
1982). Winter is defined from November to April in the Northern
Hemisphere and May to October in the Southern Hemisphere. |
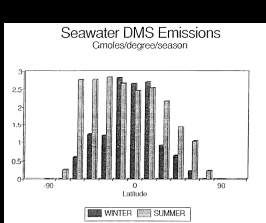
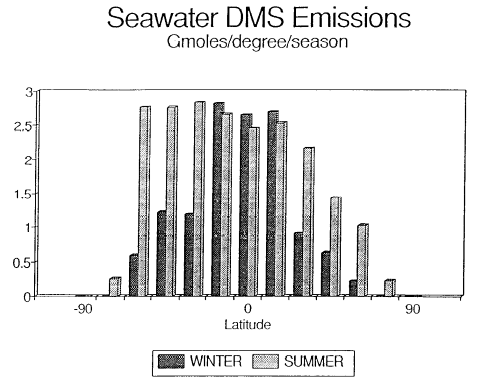
Fig. 1. Biogenic emissions of oceanic DMS to the atmosphere corresponding
to the regions shown in Table 1. The equatorial regions have been combined (5°N
to 5°S). The data have been plotted per degree of latitude to demonstrate the
large source strength in the temperate latitudes of the Southern Hemisphere.
The oceanic DMS flux of 0.5 ± 0.33 Tmol/a was obtained from extrapolation of
regional and seasonal emissions for the North Pacific Ocean (Bates et al., 1987). These emissions also can be extrapolated regionally
and seasonally to the other oceans to obtain a map of oceanic DMS emissions
(Table 1 and Figure
1). For the purposes of this analysis, the year has been divided into two
seasons, summer and winter, with the midpoints of the seasons offset from the
solstices by two months to account for the lag in ocean/soil warming. The globe
has been divided into 12 latitude belts (Table
1) after Bates
and coworkers (1987) with the areas poleward of 80° being neglected due
to snow or ice cover. For simplicity, the coastal ocean has not been separated
from the open ocean. Although DMS concentrations along the coast can be appreciably
higher than the open ocean with significant local impacts in regions with large
continental shelves (Turner et al., 1988; Turner et al., 1989; Leck
and Rodhe, 1990), the area of the biologically productive coastal zone with
higher DMS flux is generally insignificant on a regional basis (Bates et al., 1990). However, the average fluxes computed here likely underestimate
the high point source emissions from coccolithophorid blooms found in the North
Atlantic Ocean (Keller et al., 1989).
2.1.3. Oceanic emissions of non-volatile sulfur species. Sea-salt
particles are ejected into the atmosphere from bubbles bursting at the sea surface (Blanchard
and Woodcock, 1957). The magnitude of this flux is largely a function of
wind speed (Patterson et al., 1980) and the height at which the flux is calculated (Blanchard,
1985). Measurements show that the mass concentration of sea-salt particles
falls off rapidly with increasing height in the lower marine boundary layer,
dropping by a factor of two between 20 and 30 m (Blanchard et al., 1984). The number population decreases even more rapidly,
falling by three orders of magnitude between the lower marine boundary layer
and the overlying free troposphere at approximately 2 km (Patterson et al., 1980). Typical number populations of coarse (D > 1.0 µm) sea-salt particles over the open ocean at cloud height are generally
<1.0 cm (Hobbs,
1971; Patterson et al., 1980; Andreae et al., 1986) as opposed to several hundred cm for submicrometer non-sea-salt sulfate particles (Bigg et al., 1984). Consequently, although a sea-salt sulfur production
rate of 4.1 to 8.6 Tmol/a is calculated for the layer 10 to 20 m above the sea
surface (Varhelyi
and Gravenhorst, 1983; Blanchard,
1985), this sulfur has a relatively short atmospheric life-time and is not
mixed throughout the marine boundary layer. The uncertainties associated with
sea-salt emissions are difficult to estimate since the calculations are based
on rainfall rates, dry deposition rates, and aerosol concentrations that are
a strong function of wind speed and height above the sea surface.
(Hobbs,
1971; Patterson et al., 1980; Andreae et al., 1986) as opposed to several hundred cm for submicrometer non-sea-salt sulfate particles (Bigg et al., 1984). Consequently, although a sea-salt sulfur production
rate of 4.1 to 8.6 Tmol/a is calculated for the layer 10 to 20 m above the sea
surface (Varhelyi
and Gravenhorst, 1983; Blanchard,
1985), this sulfur has a relatively short atmospheric life-time and is not
mixed throughout the marine boundary layer. The uncertainties associated with
sea-salt emissions are difficult to estimate since the calculations are based
on rainfall rates, dry deposition rates, and aerosol concentrations that are
a strong function of wind speed and height above the sea surface.
2.2. Terrestrial Biogenic
2.2.1. Emission measurements. Terrestrial biogenic emissions
are extremely difficult to quantify because they include a number of sulfur
species (e.g. HS, OCS, CS,
DMS, and DMDS) and varied sources (e.g. wetlands, soils and vegetation). Unlike
oceanic fluxes, which are calculated from concentration measurements and air-sea
exchange models, terrestrial fluxes are measured directly using either enclosure
or micro-meteorological techniques. Both techniques have their limitations.
The more commonly used enclosure technique can disturb the natural habitat (e.g.
damage roots or vegetation, increase the temperature in the enclosure) and cause
high and erratic sulfur fluxes (Dacey et al., 1987). Many early measurements which reported extremely high
fluxes undoubtedly suffered from these problems. Micro-meteorological techniques
require a sulfur gas gradient in the atmosphere and eddy correlation measurements
of some other parameter (generally water vapor) since species specific sulfur
detectors currently do not have a sufficiently fast response to directly obtain
sulfur fluxes. Hence, these measurements are subject to extremely high uncertainties.
The first estimates of terrestrial biogenic sulfur emissions, based on measured
fluxes, were done by Adams
and coworkers (1981). Since then there have been many studies conducted
using increasingly better measurement technology. Recently, Lamb
and coworkers (1987) have compiled an extensive data base of over 300 field
measurements of natural sulfur emission rates from a wide range of sources at
sites in Iowa, Ohio, North Carolina, Washington and Idaho. Bare Mollisol, Histosol,
and marshland soils were sampled in addition to surface areas with row crops
(celery, carrots and onions) or natural vegetation. Above ground emissions were
also measured from agricultural crops (soybean, corn, and alfalfa) and forest
canopies (ash, oak, maple, hickory, and pine). The biogenic sulfur emission
rates measured by Lamb
and coworkers (1987) were from two (for HS)
to one (other sulfur compounds) order of magnitude lower than measurements reported
by Adams
and coworkers (1981) at the same sites. Independent data collected simultaneously
at the same sites by MacTaggart
and coworkers (1987) and Goldan
and coworkers (1987) are within a factor of two of the emission rates measured
by Lamb
and coworkers (1987). Other recently published data (Jorgensen
and Okholm-Hansen, 1985; Carroll et al., 1986; Cooper et al., 1987a; Cooper et al., 1987b; de
Mello et al., 1987; Andreae
and Andreae, 1988; Fall et al., 1988; Staubes et al., 1989) are also within the range of these emission rate estimates.
Biogenic sulfur emission measurements now extend from high latitude tundra sites
(0.083 µmol/m /d for a 150-day season, Hines
and Morrison, 1989) to the tropics (see below, Andreae
and Andreae, 1988). The various biogenic sources are reviewed below.
/d for a 150-day season, Hines
and Morrison, 1989) to the tropics (see below, Andreae
and Andreae, 1988). The various biogenic sources are reviewed below.
2.2.1.1. Marsh and tideland emissions. Wetlands are a major source
of both DMS and HS to the atmosphere.
DMS is the dominant species from both wet and dry vegetated sites and its emission
is strongly dependent on temperature (de
Mello et al., 1987) and the abundance of one species, Spartina
alterniflora (Dacey et al., 1987). HS emissions
from sediments can vary by over 5 orders of magnitude (0.045 to 4500 µmol/m˛/d)
and generally can be correlated to tidal cycling (Jorgensen
and Okholm-Hansen, 1985; Steudler
and Peterson, 1985; Carroll et al., 1986; Cooper et al., 1987a; Cooper et al., 1987b; de
Mello et al., 1987). Ninety percent of the emissions can occur in
less than 10% of the tidal cycle in a narrow region at the water's edge as the
tide rises and falls (Cooper et al., 1987a). Both the species dependency of DMS emissions and
the large variations in HS emissions
make it difficult to develop an emission rate algorithm for wetland emissions.
2.2.1.2. Freshwater emissions. DMS is the major volatile sulfur
compound in oxic freshwater lakes (Turner
and Liss, 1985), although the concentrations are much lower than that found
in seawater. Nriagu
and Holdway (1989) measured DMS in the Great Lakes, USA, and found surface
concentrations ranging from 0.1 to 1 nmol/L during the summer. The one exception
was a sample containing remnants of a diatom bloom which had a concentration
near 15 nmol/L. The calculated mean DMS flux from the Great Lakes was 0.5 µmol/m/d (Nriagu
and Holdway, 1989), or 1/10 of the average summer temperate latitude open
ocean DMS flux (Bates et al., 1987).
2.2.1.3. Soil emissions. Laboratory studies have indicated that
the sulfur flux from soils is not dependent on soil microorganisms leading to
the speculation that sulfur compounds are desorbed from soil surfaces (Goldan et al., 1987). The flux of sulfur gases from soils is exponentially
dependent on the surface soil temperature (Goldan et al., 1987) and is also a function of soil nitrogen content (Melillo
and Steudler, 1989), soil type, and moisture content (Goldan et al., 1987; Lamb et al., 1987; Staubes et al., 1989). Sulfur fluxes generally range from 0.1 to 2 µmol/m/d
for temperatures between 20 and 30°C (Goldan et al., 1987; Lamb et al., 1987; Andreae
and Andreae, 1988; Staubes et al., 1989).
2.2.1.4. Vegetation emissions. Sulfur is an essential nutrient
for all plants and is taken into the plant cells both from roots (sulfate) and
leaves (SO and other volatile sulfur
compounds) (Rennenberg, 1984). Plants are the largest sink for atmospheric OCS (Brown
and Bell, 1986; Fall et al., 1988; Mihalopoulos et al., 1989), absorbing 3 to 10 Gmol S/a through their open leaf
stomata (Goldan et al., 1988). Laboratory studies have demonstrated that OCS uptake
by plants is light dependent (Fall et al., 1988). The emission of reduced sulfur gases from plants is
also light dependent (Fall et al., 1988) and may be a mechanism by which plants dispose of excess
sulfur (Filner et al., 1984; Rennenberg,
1984). Laboratory studies have demonstrated that DMS is the dominant compound
emitted from crops (corn, alfalfa, wheat) with the flux increasing exponentially
with temperature (Fall et al., 1988). Similar results have been found in the field (Goldan et al., 1987; Lamb et al., 1987). This temperature dependence results in a significant
seasonal cycle in biogenic sulfur emissions at higher latitudes. Measurements
in the tropics, however, show no significant differences between the wet and
dry seasons (Andreae et al., 1990). Sulfur emissions from forests and croplands generally
range from 0.4 to 4 µmol/m/d for temperatures
between 20 and 30°C (Goldan et al., 1987; Lamb et al., 1987; Andreae
and Andreae, 1988). The emissions calculated below are a gross flux as measured
by enclosure techniques and hence do not include OCS uptake by plants.
2.2.2. Emission algorithms. A high degree of variability in local
natural terrestrial sulfur emissions results in large uncertainties when extrapolating
to regional and global scales. While the mechanisms controlling the release
of natural sulfur emissions are not well understood, field observations have
demonstrated that temperature plays a dominant role and that characteristic
temperature-flux patterns can be determined for individual compounds and sources.
As temperatures fall below 0°C, sulfur emissions fall below the lower detection
limit of the methods used by Lamb
and coworkers (1987) which is approximately 45 nmol/m/d
for surface fluxes and 1.8 nmol/m/d for vegetative
emissions. The low emission rate below 0°C is not unexpected considering the
minimal biological enzymatic activity which occurs at those temperatures (Tauber,
1949). Sulfur emissions tend to increase logarithmically with increasing
temperature for normal ambient temperatures (10°C to 35°C) (Goldan et al., 1987; Lamb et al., 1987). Although biological enzymatic activity normally reaches
a plateau at high temperatures, a saturation point is not always apparent in
field measurements. This may be the result of temporarily increased emissions
which result from the stress experienced by the vegetation enclosed within a
chamber at high temperatures (above 35°C).
The emission characteristics observed in field studies suggest that the following
form of the Michaelis-Menten equation can be used to mathematically represent
the relationship between ambient temperatures and natural sulfur emissions:
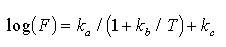 (2)
(2)
where F is the sulfur emission rate ( µmol/m/d). T is ambient temperature (°C), k and k
and k are rate constants
determined by a non-linear least squares fit to emission rate data, and k
are rate constants
determined by a non-linear least squares fit to emission rate data, and k is a rate constant determined by the lower detection limit. Equation (2) provides
an emission algorithm that predicts very low emissions near 0°C, a logarithmic
increase in emissions at intermediate temperatures, and approaches a maximum
emission rate at some saturation temperature. Guenther and coworkers (1989)
have applied a non-linear least squares best fit technique to the emissions
data collected by Lamb
and coworkers (1987) to estimate k and k for OCS, CS,
DMDS, DMS and HS for five surface
categories which include wetland soils, organic soils, other soils, water, and
agricultural crops (other than corn) and three vegetation categories which are
deciduous and coniferous forest canopies and corn biomass. Additional natural
sulfur compounds (e.g. mercaptans) are typically released at rates which are
small (<1%) relative to the total sulfur flux.
is a rate constant determined by the lower detection limit. Equation (2) provides
an emission algorithm that predicts very low emissions near 0°C, a logarithmic
increase in emissions at intermediate temperatures, and approaches a maximum
emission rate at some saturation temperature. Guenther and coworkers (1989)
have applied a non-linear least squares best fit technique to the emissions
data collected by Lamb
and coworkers (1987) to estimate k and k for OCS, CS,
DMDS, DMS and HS for five surface
categories which include wetland soils, organic soils, other soils, water, and
agricultural crops (other than corn) and three vegetation categories which are
deciduous and coniferous forest canopies and corn biomass. Additional natural
sulfur compounds (e.g. mercaptans) are typically released at rates which are
small (<1%) relative to the total sulfur flux.
The temperature dependent algorithms used to predict natural sulfur emissions
do not account for all of the variation in observed emissions. Other important
environmental parameters may include, but are not limited to, tidal flushing,
availability of sulfur, soil moisture, soil pH, mineral composition, ground
cover, and solar radiation. A more accurate estimation of biogenic sulfur emissions
requires a better understanding of the factors which influence natural emissions
and the means to extrapolate any additional parameters which are determined
to be important.
2.2.3. Global inventory of terrestrial biogenic volatile sulfur emissions.
Initial attempts to describe global biogeochemical sulfur circulation indirectly
estimated terrestrial biogenic sulfur emissions by calculating the amount necessary
to balance the global sulfur budget. Global average terrestrial emissions of
3.3 µmol/m/d (Friend, 1973) to 63 µmol/m/d (Eriksson,
1963) were required to balance global sulfur budgets. As a first step towards
extrapolating actual terrestrial emission rate measurements to the global scale,
the biogenic sulfur emissions algorithms developed by Guenther
and coworkers (1989) are used here to generate a global emissions inventory.
Climatic data compiled on a 2° latitude by 2° longitude spatial scale and monthly
temperature scale by Shea
(1986) and land cover data compiled by Wilson
and Henderson-Sellers (1985) on a 1° latitude by 1° longitude scale provide
the temperature and source inputs required for a global inventory. The primary
(>50% of the grid) and secondary (25% to 50% of the grid) land cover types
were compiled into 12 simple surface types using the types and proportions given
in Table 12 of Henderson-Sellers
and coworkers (1986). The areas associated with each of these 12 surface
types were summed by latitude band and these sums were used as the primary input
into the emission calculation. It was assumed that primary vegetation covered
75% of a grid and secondary vegetation covered 25% of the grid in this summation
process. Areas of open water and ice were eliminated from the summation process.
For this preliminary estimate, the earth land mass was divided into the same
latitudinal regions and seasons used for estimating oceanic sulfur emissions.
Seasonal emission rates were calculated for five sources: cropland vegetation,
wetlands, deciduous and coniferous canopies, and soils (total soil area = natural
vegetation area + agricultural land areas + bare soil area).
If your browser cannot view the following table correctly,
click this link for a GIF image of Table 2.
TABLE 2. Terrestrial sulfur fluxes
|
|
winter |
summer |
winter |
summer |
|
emiss |
emiss |
flux |
flux |
| Region |
--(µmol S/m˛/d)-- |
--(10 mol/d)-- |
|
| 80°–65°N |
0.00 |
0.01 |
0.0 |
0.1 |
| 65°–50°N |
0.00 |
0.10 |
0.0 |
2.2 |
| 50°–35°N |
0.02 |
0.19 |
0.6 |
4.6 |
| 35°–20°N |
0.16 |
0.33 |
3.6 |
7.6 |
| 20°–5°N |
0.39 |
0.43 |
6.6 |
7.3 |
| 5°N–0° |
0.55 |
0.53 |
3.2 |
3.1 |
| 0°–5°S |
0.55 |
0.56 |
3.0 |
3.1 |
| 5°–20°S |
0.36 |
0.43 |
5.3 |
6.3 |
| 20°–35°S |
0.15 |
0.25 |
1.8 |
2.9 |
| 35°–50°S |
0.09 |
0.15 |
0.2 |
0.3 |
| 50°–65°S |
0.03 |
0.07 |
0.0 |
0.0 |
| 65°–80°S |
0.00 |
0.00 |
0.0 |
0.0 |
| N. Hemisphere |
13.9 |
24.9 |
| S. Hemisphere |
10.2 |
12.6 |
|
| Winter is defined from November to April in the Northern |
| Hemisphere and May to October in the Southern Hemisphere. |
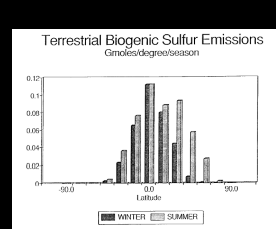
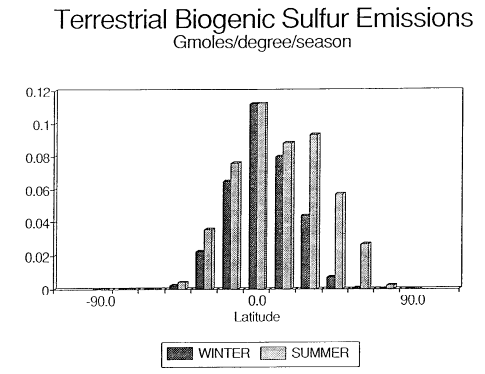
Fig. 2. Biogenic emissions of terrestrial sulfur compounds to the atmosphere.
The data have been plotted per degree of latitude to demonstrate the uneven
distribution between the northern and southern hemispheres.
The resulting total annual global emission estimate of 0.011 Tmol is equivalent
to 0.21 µmol/m/d which is only slightly higher
than the estimated U.S. national average emission rate estimate of 0.18 µmol/m/d (Guenther et al., 1989). The tropical regions (20°N to 20°S) generate 61% of
all emissions (Table 2 and Figure 2). Because of the temperature dependence
of the emission algorithms, the summertime (May to October in the Northern Hemisphere,
November to April in the Southern Hemisphere) emissions contribute 61% of the
global annual flux. Seasonal emissions are nearly constant in the equatorial
region. The predicted emissions are primarily OCS (47%), DMS (27%), and HS
(20%). CS and DMDS each contribute
about 3% on a global scale but as much as 10% in individual regions (Table
3). Emissions estimated for the northern hemisphere are 70% greater than
those predicted for the southern hemisphere. Emissions from deciduous canopies
dominate (58%) the total emission rate with soils (29%) and coniferous canopies
(9%) contributing the majority of the remainder (Table
4). Coniferous canopies are important (>27%) at high latitudes (>35°N
or S) and predominate in the northern boreal biome. Croplands and wetlands together
account for 4% of the annual global flux. The areas used to calculate the emissions
by source are shown in Table
5.
If your browser cannot view the following table correctly, click this link for a GIF image of Table
3.
TABLE 3. Terrestrial sulfur fluxes by compound
|
|
OCSą |
CS |
DMDS |
DMS |
HS |
| Region |
--(10 mol/d)-- |
|
| 80°–65°N |
0.03 |
0.01 |
0.01 |
0.01 |
0.00 |
| 65°–50°N |
0.65 |
0.07 |
0.05 |
0.16 |
0.18 |
| 50°–35°N |
1.35 |
0.13 |
0.09 |
0.57 |
0.46 |
| 35°–20°N |
2.48 |
0.22 |
0.15 |
1.68 |
1.08 |
| 20°–5°N |
3.03 |
0.23 |
0.15 |
2.17 |
1.34 |
| 5°N–0° |
1.51 |
0.09 |
0.06 |
0.80 |
0.66 |
| 0°–5°S |
1.43 |
0.09 |
0.06 |
0.79 |
0.63 |
| 5°–20°S |
2.82 |
0.19 |
0.13 |
1.45 |
1.20 |
| 20°–35°S |
1.15 |
0.09 |
0.07 |
0.59 |
0.46 |
| 35°–50°S |
0.13 |
0.01 |
0.01 |
0.04 |
0.04 |
| 50°–65°S |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
| 65°–80°S |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
| N. Hemisphere |
9.05 |
0.75 |
0.51 |
5.40 |
3.72 |
| S. Hemisphere |
5.54 |
0.38 |
0.27 |
2.86 |
2.33 |
|
| ą Fluxes are a gross flux as measured in enclosures
and hence do not take into |
| account OCS uptake by vegetation. 82%
of the flux listed above is from natural |
| vegetation and crops. |
If your browser cannot view the following table correctly,
click this link for a GIF image of Table 4.
TABLE 4. Terrestrial sulfur fluxes by source
|
|
conifers |
deciduous |
crops |
soils |
wetlands |
| Region |
--(10 mol/d)-- |
|
| 80°–65°N |
0.02 |
0.02 |
0.00 |
0.02 |
0.00 |
| 65°–50°N |
0.49 |
0.32 |
0.05 |
0.22 |
0.02 |
| 50°–35°N |
0.69 |
0.97 |
0.15 |
0.69 |
0.10 |
| 35°–20°N |
0.51 |
2.52 |
0.15 |
2.11 |
0.30 |
| 20°–5°N |
0.44 |
3.95 |
0.07 |
2.30 |
0.15 |
| 5°N–0° |
0.08 |
2.36 |
0.00 |
0.66 |
0.02 |
| 0°–5°S |
0.07 |
2.24 |
0.00 |
0.65 |
0.04 |
| 5°–20°S |
0.32 |
4.01 |
0.00 |
1.39 |
0.06 |
| 20°–35°S |
0.21 |
1.36 |
0.02 |
0.76 |
0.01 |
| 35°–50°S |
0.06 |
0.12 |
0.00 |
0.05 |
0.00 |
| 50°–65°S |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
| 65°–80°S |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
| N. Hemisphere |
2.24 |
10.15 |
0.43 |
6.01 |
0.59 |
| S. Hemisphere |
0.67 |
7.73 |
0.03 |
2.85 |
0.11 |
|
If your browser cannot view the following table correctly,
click this link for a GIF image of Table 5.
TABLE 5. Terrestrial areas by source (areas of open water and ice are
eliminated from totals)
|
|
Total |
Natural |
Soil |
Agriculture |
Wetland |
|
Area |
Vegetation |
Area |
Area |
Area |
| Region |
--(10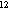 m˛)-- m˛)-- |
|
| 80°–65°N |
9.8 |
9.0 |
9.5 |
0.0 |
0.3 |
| 65°–50°N |
21.6 |
15.5 |
20.5 |
2.7 |
1.0 |
| 50°–35°N |
24.5 |
17.7 |
23.2 |
4.7 |
1.3 |
| 35°–20°N |
23.1 |
19.2 |
22.4 |
2.7 |
0.7 |
| 20°–5°N |
16.8 |
14.7 |
16.6 |
1.0 |
0.2 |
| 5°N–0° |
5.8 |
5.5 |
5.7 |
0.0 |
0.0 |
| 0°–5°S |
5.4 |
5.1 |
5.4 |
0.1 |
0.1 |
| 5°–20°S |
14.6 |
13.4 |
14.5 |
0.1 |
0.1 |
| 20°–35°S |
11.6 |
10.9 |
11.6 |
0.4 |
0.1 |
| 35°–50°S |
1.9 |
1.8 |
1.9 |
0.1 |
0.0 |
| 50°–65°S |
0.1 |
0.1 |
0.1 |
0.0 |
0.0 |
| 65°–80°S |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
| N. Hemisphere |
101.5 |
81.6 |
98.1 |
11.1 |
3.4 |
| S. Hemisphere |
33.7 |
31.3 |
33.5 |
0.6 |
0.3 |
|
The global terrestrial emission inventory described in this paper is a conservative
estimate. A low sulfur emission rate, representative of alfalfa and row crops,
was applied to all croplands. Cropland emissions may be considerably higher
if significant portions of croplands contain plants, such as corn, which have
much higher sulfur emission rates. If, for example, 25% of the world's croplands
were corn (or crops with similar sulfur emission rates) then the estimated annual
sulfur emission rate for croplands would increase from 1.6 × 10 to 1.8 × 10
to 1.8 × 10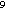 moles. Similarly, a low emission
rate was applied to all soils other than wetlands. If 10% of the world's soils
emit sulfur at a rate similar to the organic histosol soils measured by Lamb
and coworkers (1987), then the estimated annual sulfur emission rate for
non-wetland soils would increase from 3.4 × 10 to 6.9 × 10 moles. Because of the exponential
relationship between temperature and emissions, higher emissions would also
be calculated if smaller spatial or temporal resolution were used. For example, Guenther
and coworkers (1989) have estimated that using monthly average instead of
daily average temperatures underestimates biogenic sulfur emissions by up to
25%. Another simplification in the methodology used to calculate this global
inventory, the use of a constant peak biomass, overestimates emissions by assuming
that all biomass is present at all times while in reality deciduous canopies
and croplands have very little biomass at certain times of the year. The importance
of this overestimated biomass is probably minimal since cold temperatures, resulting
in a very low biomass emission rate, correspond with the periods of overestimated
biomass.
moles. Similarly, a low emission
rate was applied to all soils other than wetlands. If 10% of the world's soils
emit sulfur at a rate similar to the organic histosol soils measured by Lamb
and coworkers (1987), then the estimated annual sulfur emission rate for
non-wetland soils would increase from 3.4 × 10 to 6.9 × 10 moles. Because of the exponential
relationship between temperature and emissions, higher emissions would also
be calculated if smaller spatial or temporal resolution were used. For example, Guenther
and coworkers (1989) have estimated that using monthly average instead of
daily average temperatures underestimates biogenic sulfur emissions by up to
25%. Another simplification in the methodology used to calculate this global
inventory, the use of a constant peak biomass, overestimates emissions by assuming
that all biomass is present at all times while in reality deciduous canopies
and croplands have very little biomass at certain times of the year. The importance
of this overestimated biomass is probably minimal since cold temperatures, resulting
in a very low biomass emission rate, correspond with the periods of overestimated
biomass.
It is possible that an improved inventory methodology (e.g. better resolution
and more specific source types) would result in a global emission rate estimate
that is as much as a factor of two greater than our preliminary estimate of
0.011 Tmol/a. This would still be one or two orders of magnitude less than the
0.16 to 3.4 Tmol/a required to balance the early global sulfur budgets (e.g.
Eriksson, 1963; Friend,
1973; Granat et al., 1976).
2.3. Volcanic
Sulfur gases are emitted to the atmosphere from both erupting and non-erupting
degassing volcanos with the more violent eruptions constituting the major source
of sulfur to the stratosphere (Pinto et al., 1989). The major sulfur compound emitted from volcanos is
SO with SO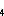
 and HS generally comprising less
than 1% (Stoiber et al., 1987) and OCS less than 0.1% (Khalil
and Rasmussen, 1984; Belviso et al., 1986) of the total sulfur emissions. The most recent estimate
of annual SO emissions to the atmosphere
by volcanos is 0.29 Tmol/a (Stoiber et al., 1987). This estimate is based on an extrapolation of direct
measurements of volcanic SO and
include 0.11 Tmol/a from 102 degassing volcanos and 0.18 Tmol/a from approximately
60 erupting volcanos. The estimate of the number of volcanos is an average over
the past 400 years.
and HS generally comprising less
than 1% (Stoiber et al., 1987) and OCS less than 0.1% (Khalil
and Rasmussen, 1984; Belviso et al., 1986) of the total sulfur emissions. The most recent estimate
of annual SO emissions to the atmosphere
by volcanos is 0.29 Tmol/a (Stoiber et al., 1987). This estimate is based on an extrapolation of direct
measurements of volcanic SO and
include 0.11 Tmol/a from 102 degassing volcanos and 0.18 Tmol/a from approximately
60 erupting volcanos. The estimate of the number of volcanos is an average over
the past 400 years.
Not all of this sulfur is emitted to the troposphere. Volcanoes with a Volcanic
Explosivity Index (VEI) of 4 or greater produce approximately 7% of the total
sulfur emitted from volcanoes (Stoiber et al., 1987) and have eruption cloud columns which can reach the
stratosphere. During the period 1975-1985 seven of the eight volcanoes with
VEI of four or greater were located in the northern hemisphere (McClelland et al., 1989) and are evident in the stratospheric aerosol mixing
ratio (Hofmann,
1990).
Unlike biogenic sulfur emissions which have relatively small emission rates
over large areas, volcanism is concentrated in small segments of the Earth's
crust, generally related to plate boundaries. Two-thirds of the world's known
volcanos are in the northern hemisphere and only 18% are between 10°S and the
South Pole (Simkin et al., 1981; Simkin
and Siebert, 1984). The emission rate of SO from any one volcano is episodic and varies with eruptive activity (Malinconico,
1987), making it extremely difficult to calculate the uncertainty of the
estimates.
2.4. Other Natural Sources
2.4.1. Biomass Burning. The burning of forests, grasslands, and
agricultural wastes can not necessarily be considered a "natural"
sulfur emission since as much as 95% of global burning is thought to be human
initiated (Hileman,
1990). However, biomass burning is a significant source of SO to the atmosphere. Field measurements of sulfur gases and sulfate aerosol particles
have been made in prescribed burns of chaparral and brush near Los Angeles,
California (Hegg et al., 1987) and in the Amazon basin (Andreae et al., 1988). The molar ratio of total sulfur emissions to CO emissions ranged from 0.00021 (Hegg et al., 1987) to 0.00032 (Andreae et al., 1988). These emission ratios can be used with global estimates
of CO emissions from biomass burning (Seiler
and Crutzen, 1980; Hao et al., 1989; Dignon
and Penner, 1990; Hileman,
1990; Logan et al., 1990) to calculate a total sulfur emission of 0.045 to 0.092
Tmol S/a. Based on the sulfur content of dry matter before and after a fire, Delmas
and Servant (1988) calculated an emission of 0.11 Tmol S/a from biomass
burning, a value very similar to that calculated here. Delmas
(1982) proposed a range of 0.013 to 0.038 Tmol S/a for total annual emissions
from savanna fires. Dignon
and Penner (1990) have estimated that savanna fires may account for 50 to
60 percent of global biomass burning emissions. Thus, the total sulfur source
based on the savanna estimates of Delmas
(1982) would yield roughly 0.026 to 0.063 Tmol S/a. The estimates presented
by Delmas
(1982) and later by Delmas
and Servant (1988) are similar to the range presented here.
Most of the burning occurs during the dry season in the tropics with roughly
80 percent of the emissions between 25°N and 25°S (Dignon
and Penner, 1990). Based on the work of Hao et al. (1989) and Lacey et al. (1982) it is estimated that less than 20 percent of the biomass
burning in the southern hemisphere occurs in summer. In the northern hemisphere,
tropical burning is predominantly in winter while at higher latitudes, summer
is the dominant burning season. Since most burning area estimates are known
to no better than ±50% (Robinson,
1989), and there are few emission measurements for sulfur species, these
estimates are expected to have a considerable uncertainty. The only aspect of
biomass burning that is certain at this time, is that the amount of burning
has accelerated greatly in the past few decades with detrimental environmental
consequences (Hileman,
1990).
2.4.2. Aeolian dust. Massive quantities of dust are transported
throughout the troposphere due to wind blown erosion. Although the dust carried
across the tropical North Atlantic Ocean from the Sahara desert is associated
with approximately 60% of the mean total concentration of sulfate reaching Barbados,
this sulfate is most likely associated with anthropogenic as opposed to crustal
Saharan material (Savoie et al., 1989). Like sea-salt-sulfate, the sulfur derived from soils
is principally contained on larger (>1 µm) particles.
Go back to previous section or forward to next section
Return to Abstract
PMEL Outstanding Papers
PMEL Publications Search
PMEL Homepage