Human activity is rapidly changing the composition of the earth's atmosphere,
contributing to warming from excess carbon dioxide (CO![]() )
along with other trace gases such as water vapor, chlorofluorocarbons, methane
and nitrous oxide. These anthropogenic "greenhouse gases" play a
critical role in controlling the earth's climate because they increase the
infrared opacity of the atmosphere, causing the surface of the planet to warm.
The release of CO
)
along with other trace gases such as water vapor, chlorofluorocarbons, methane
and nitrous oxide. These anthropogenic "greenhouse gases" play a
critical role in controlling the earth's climate because they increase the
infrared opacity of the atmosphere, causing the surface of the planet to warm.
The release of CO![]() from
fossil fuel consumption or the burning of forests for farming or pasture contributes
approximately 7 petagrams of carbon (1 Pg C = 1 × 10
from
fossil fuel consumption or the burning of forests for farming or pasture contributes
approximately 7 petagrams of carbon (1 Pg C = 1 × 10![]() g
C) to the atmosphere each year. Approximately 3 Pg C of this "anthropogenic
CO
g
C) to the atmosphere each year. Approximately 3 Pg C of this "anthropogenic
CO![]() " accumulates
in the atmosphere annually, and the remaining 4 Pg C is stored in the terrestrial
biosphere and the ocean.
" accumulates
in the atmosphere annually, and the remaining 4 Pg C is stored in the terrestrial
biosphere and the ocean.
Where and how land and ocean regions vary in their uptake of CO![]() from
year to year is the subject of much scientific research and debate. Future
decisions on regulating emissions of greenhouse gases should be based on more
accurate models of the global cycling of carbon and the regional sources and
sinks for anthropogenic CO
from
year to year is the subject of much scientific research and debate. Future
decisions on regulating emissions of greenhouse gases should be based on more
accurate models of the global cycling of carbon and the regional sources and
sinks for anthropogenic CO![]() ,
models that have been adequately tested against a well-designed system of measurements.
The construction of a believable present-day carbon budget is essential for
the reliable prediction of changes in atmospheric CO
,
models that have been adequately tested against a well-designed system of measurements.
The construction of a believable present-day carbon budget is essential for
the reliable prediction of changes in atmospheric CO![]() and
global temperatures from available emissions scenarios.
and
global temperatures from available emissions scenarios.
The ocean plays a critical role in the global carbon cycle as a vast reservoir
that exchanges carbon rapidly with the atmosphere, and takes up a substantial
portion of anthropogenically-released carbon from the atmosphere. A significant
impetus for carbon cycle research over the past several decades has been to
achieve a better understanding of the ocean's role as a sink for anthropogenic
CO![]() . There are
only three global reservoirs with exchange rates fast enough to vary significantly
on the scale of decades to centuries: the atmosphere, the terrestrial biosphere
and the ocean. Approximately 93% of the carbon is located in the ocean, which
is able to hold much more carbon than the other reservoirs because most of
the CO
. There are
only three global reservoirs with exchange rates fast enough to vary significantly
on the scale of decades to centuries: the atmosphere, the terrestrial biosphere
and the ocean. Approximately 93% of the carbon is located in the ocean, which
is able to hold much more carbon than the other reservoirs because most of
the CO![]() that
diffuses into the oceans reacts with seawater to form carbonic acid and its
dissociation products, bicarbonate and carbonate ions (Figure
1).
that
diffuses into the oceans reacts with seawater to form carbonic acid and its
dissociation products, bicarbonate and carbonate ions (Figure
1).
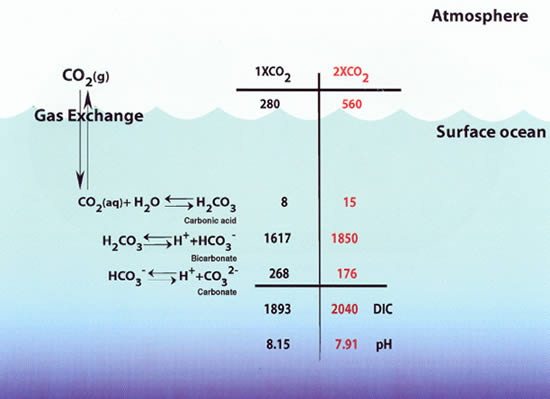
Figure 1. Schematic diagram of the carbon dioxide (CO![]() )
system in seawater. The 1 × CO
)
system in seawater. The 1 × CO![]() concentrations
are for a surface ocean in equilibrium with a pre-industrial atmospheric
CO
concentrations
are for a surface ocean in equilibrium with a pre-industrial atmospheric
CO![]() level of
280 ppm. The 2 × CO
level of
280 ppm. The 2 × CO![]() concentrations
are for a surface ocean in equilibrium with an atmospheric CO
concentrations
are for a surface ocean in equilibrium with an atmospheric CO![]() level
of 560 ppm. Current model projections indicate that this level could be reached
sometime in the second half of this century. The atmospheric values are in
units of ppm. The oceanic concentrations, which are for the surface mixed
layer, are in units of µmol kg
level
of 560 ppm. Current model projections indicate that this level could be reached
sometime in the second half of this century. The atmospheric values are in
units of ppm. The oceanic concentrations, which are for the surface mixed
layer, are in units of µmol kg![]() .
.
Our present understanding of the temporal and spatial distribution of net
CO![]() flux into
or out of the ocean is derived from a combination of field data, which is limited
by sparse temporal and spatial coverage, and model results, which are validated
by comparisons with the observed distributions of tracers, including natural
carbon-14 (
flux into
or out of the ocean is derived from a combination of field data, which is limited
by sparse temporal and spatial coverage, and model results, which are validated
by comparisons with the observed distributions of tracers, including natural
carbon-14 (![]() C),
and anthropogenic chlorofluorocarbons, tritium (
C),
and anthropogenic chlorofluorocarbons, tritium (![]() H)
and bomb
H)
and bomb ![]() C.
The latter two radioactive tracers were introduced into the atmosphere-ocean
system by atomic testing in the mid 20th century. With additional data from
the recent global survey of CO
C.
The latter two radioactive tracers were introduced into the atmosphere-ocean
system by atomic testing in the mid 20th century. With additional data from
the recent global survey of CO![]() in
the ocean (1991–1998), carried out cooperatively as part of the Joint
Global Ocean Flux Study (JGOFS) and the World Ocean Circulation Experiment
(WOCE) Hydrographic Program, it is now possible to characterize in a quantitative
way the regional uptake and release of CO
in
the ocean (1991–1998), carried out cooperatively as part of the Joint
Global Ocean Flux Study (JGOFS) and the World Ocean Circulation Experiment
(WOCE) Hydrographic Program, it is now possible to characterize in a quantitative
way the regional uptake and release of CO![]() and
its transport in the ocean. In this paper, we summarize our present understanding
of the exchange of CO
and
its transport in the ocean. In this paper, we summarize our present understanding
of the exchange of CO![]() across
the air-sea interface and the storage of natural and anthropogenic CO
across
the air-sea interface and the storage of natural and anthropogenic CO![]() in
the ocean's interior.
in
the ocean's interior.
The history of large-scale CO![]() observations
in the ocean date back to the 1970s and 1980s. Measurements of the partial
pressure of CO
observations
in the ocean date back to the 1970s and 1980s. Measurements of the partial
pressure of CO![]() (pCO
(pCO![]() ),
total dissolved inorganic carbon (DIC) and total alkalinity (A
),
total dissolved inorganic carbon (DIC) and total alkalinity (A![]() )
were made during the global Geochemical Ocean Sections (GEOSECS) expeditions
between 1972 and 1978, the Transient Tracers in the Oceans (TTO) North Atlantic
and Tropical Atlantic Surveys in 1981–83, the South Atlantic Ventilation
Experiment (SAVE) from 1988–1989, the French Southwest Indian Ocean experiment,
and numerous other smaller expeditions in the Pacific and Indian Oceans in
the 1980s. These studies provided marine chemists with their first view of
the carbon system in the global ocean.
)
were made during the global Geochemical Ocean Sections (GEOSECS) expeditions
between 1972 and 1978, the Transient Tracers in the Oceans (TTO) North Atlantic
and Tropical Atlantic Surveys in 1981–83, the South Atlantic Ventilation
Experiment (SAVE) from 1988–1989, the French Southwest Indian Ocean experiment,
and numerous other smaller expeditions in the Pacific and Indian Oceans in
the 1980s. These studies provided marine chemists with their first view of
the carbon system in the global ocean.
These data were collected at a time when no common reference materials or
standards were available. As a result, analytical differences between measurement
groups were as large as 29 µmol kg![]() for
both DIC and A
for
both DIC and A![]() ,
which corresponds to more than 1% of the ambient values. Large adjustments
had to be made for each of the data sets based on deepwater comparisons at
nearby stations before individual cruise data could be compared. These differences
were often nearly as large as the anthropogenic CO
,
which corresponds to more than 1% of the ambient values. Large adjustments
had to be made for each of the data sets based on deepwater comparisons at
nearby stations before individual cruise data could be compared. These differences
were often nearly as large as the anthropogenic CO![]() signal
that investigators were trying to determine (Gruber
et al., 1996). Nevertheless, these early data sets made up a component
of the surface ocean pCO
signal
that investigators were trying to determine (Gruber
et al., 1996). Nevertheless, these early data sets made up a component
of the surface ocean pCO![]() measurements
for a global climatology and also provided researchers with new insights into
the distribution of anthropogenic CO
measurements
for a global climatology and also provided researchers with new insights into
the distribution of anthropogenic CO![]() in
the ocean, particularly in the Atlantic Ocean.
in
the ocean, particularly in the Atlantic Ocean.
At the onset of the Global Survey of CO![]() in
the Ocean (Figure 2), several events took place
in the United States and in international CO
in
the Ocean (Figure 2), several events took place
in the United States and in international CO![]() measurement
communities that significantly improved the overall precision and accuracy
of the large-scale measurements. In the United States, the CO
measurement
communities that significantly improved the overall precision and accuracy
of the large-scale measurements. In the United States, the CO![]() measurement
program was co-funded by the Department of Energy (DOE), the National Oceanic
and Atmospheric Administration (NOAA) and the National Science Foundation (NSF)
under the technical guidance of the U.S. CO
measurement
program was co-funded by the Department of Energy (DOE), the National Oceanic
and Atmospheric Administration (NOAA) and the National Science Foundation (NSF)
under the technical guidance of the U.S. CO![]() Survey
Science Team. This group of academic and government scientists adopted and
perfected the recently developed coulometric titration method for DIC determination
that had demonstrated the capability to meet the required goals for precision
and accuracy. They advocated the development and distribution of certified
reference materials (CRMs) for DIC, and later for A
Survey
Science Team. This group of academic and government scientists adopted and
perfected the recently developed coulometric titration method for DIC determination
that had demonstrated the capability to meet the required goals for precision
and accuracy. They advocated the development and distribution of certified
reference materials (CRMs) for DIC, and later for A![]() ,
for international distribution under the direction of Andrew Dickson of Scripps
Institution of Oceanography (see sidebar). They also supported a shore-based
intercomparison experiment under the direction of Charles Keeling, also of
Scripps. Through international efforts, the development of protocols for CO
,
for international distribution under the direction of Andrew Dickson of Scripps
Institution of Oceanography (see sidebar). They also supported a shore-based
intercomparison experiment under the direction of Charles Keeling, also of
Scripps. Through international efforts, the development of protocols for CO![]() analyses
were adopted for the CO
analyses
were adopted for the CO![]() survey.
The international partnerships fostered by JGOFS resulted in several intercomparison
CO
survey.
The international partnerships fostered by JGOFS resulted in several intercomparison
CO![]() exercises
hosted by France, Japan, Germany and the United States. Through these and other
international collaborative programs, the measurement quality of the CO
exercises
hosted by France, Japan, Germany and the United States. Through these and other
international collaborative programs, the measurement quality of the CO![]() survey
data was well within the measurement goals of ±3 µmol kg
survey
data was well within the measurement goals of ±3 µmol kg![]() and ±5 µmol
kg
and ±5 µmol
kg![]() ,
respectively, for DIC and A
,
respectively, for DIC and A![]() .
.
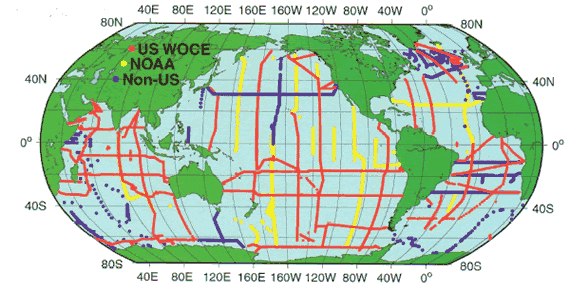
Figure 2. The Global Survey of CO![]() in
the Ocean: cruise tracks and stations occupied between 1991 and 1998.
in
the Ocean: cruise tracks and stations occupied between 1991 and 1998.
Several other developments significantly enhanced the quality of the CO![]() data
sets during this period. New methods were developed for automated underway
and discrete pCO
data
sets during this period. New methods were developed for automated underway
and discrete pCO![]() measurements.
An extremely precise method for pH measurements based on spectrophotometry
was also developed by Robert Byrne and his colleagues at the University of
South Florida. These improvements ensured that the internal consistency of
the carbonate system in seawater could be tested in the field whenever more
than two components of the carbonate system were measured at the same location
and time. This allowed several investigators to test the overall quality of
the global CO
measurements.
An extremely precise method for pH measurements based on spectrophotometry
was also developed by Robert Byrne and his colleagues at the University of
South Florida. These improvements ensured that the internal consistency of
the carbonate system in seawater could be tested in the field whenever more
than two components of the carbonate system were measured at the same location
and time. This allowed several investigators to test the overall quality of
the global CO![]() data
set based upon CO
data
set based upon CO![]() system
thermodynamics. Laboratories all around the world contributed to a very large
and internally consistent global ocean CO
system
thermodynamics. Laboratories all around the world contributed to a very large
and internally consistent global ocean CO![]() data
set determined at roughly 100,000 sample locations in the Atlantic, Pacific,
Indian and Southern oceans (Figure 2). The data
from the CO
data
set determined at roughly 100,000 sample locations in the Atlantic, Pacific,
Indian and Southern oceans (Figure 2). The data
from the CO![]() survey
are available through the Carbon Dioxide Information and Analysis Center (CDIAC)
at Oak Ridge National Laboratory as Numeric Data Packages and on the World
Wide Web (http://cdiac.esd.ornl.gov/home.html).
Taro Takahashi and his collaborators have also amassed a large database of
surface ocean pCO
survey
are available through the Carbon Dioxide Information and Analysis Center (CDIAC)
at Oak Ridge National Laboratory as Numeric Data Packages and on the World
Wide Web (http://cdiac.esd.ornl.gov/home.html).
Taro Takahashi and his collaborators have also amassed a large database of
surface ocean pCO![]() measurements,
spanning more than 30 years, into a pCO
measurements,
spanning more than 30 years, into a pCO![]() climatology
for the global ocean (Takahashi
et al., 2002). These data have been used to determine the global and
regional fluxes for CO
climatology
for the global ocean (Takahashi
et al., 2002). These data have been used to determine the global and
regional fluxes for CO![]() in
the ocean.
in
the ocean.
| Reference Materials For Oceanic CO2 Measurements |
In seawater, CO![]() molecules
are present in three major forms: the undissociated species in water, [CO
molecules
are present in three major forms: the undissociated species in water, [CO![]() ]aq,
and two ionic species, [HCO
]aq,
and two ionic species, [HCO![]()
![]() ]
and [CO
]
and [CO![]()
![]() ]
(Figure 1). The concentration of [CO
]
(Figure 1). The concentration of [CO![]() ]aq depends
upon the temperature and chemical composition of seawater. The amount of [CO
]aq depends
upon the temperature and chemical composition of seawater. The amount of [CO![]() ]aq is
proportional to the partial pressure of CO
]aq is
proportional to the partial pressure of CO![]() exerted
by seawater. The difference between the pCO
exerted
by seawater. The difference between the pCO![]() in
surface seawater and that in the overlying air represents the thermodynamic
driving potential for the CO
in
surface seawater and that in the overlying air represents the thermodynamic
driving potential for the CO![]() transfer
across the sea surface. The pCO
transfer
across the sea surface. The pCO![]() in
surface seawater is known to vary geographically and seasonally over a range
between about 150 µatm and 750 µatm, or about 60% below and 100%
above the current atmospheric pCO2 level of about 370 µatm. Since the
variation of pCO2 in the surface ocean is much greater than the atmospheric pCO
in
surface seawater is known to vary geographically and seasonally over a range
between about 150 µatm and 750 µatm, or about 60% below and 100%
above the current atmospheric pCO2 level of about 370 µatm. Since the
variation of pCO2 in the surface ocean is much greater than the atmospheric pCO![]() seasonal
variability of about 20 µatm in remote uncontaminated marine air, the
direction and magnitude of the sea-air CO
seasonal
variability of about 20 µatm in remote uncontaminated marine air, the
direction and magnitude of the sea-air CO![]() transfer
flux are regulated primarily by changes in the oceanic pCO
transfer
flux are regulated primarily by changes in the oceanic pCO![]() .
The average pCO
.
The average pCO![]() of
the global ocean is about 7 µatm lower than the atmosphere, which is
the primary driving force for uptake by the ocean (see Figure
6 in Karl et al., this issue).
of
the global ocean is about 7 µatm lower than the atmosphere, which is
the primary driving force for uptake by the ocean (see Figure
6 in Karl et al., this issue).
The pCO![]() in
mixed-layer waters that exchange CO
in
mixed-layer waters that exchange CO![]() directly
with the atmosphere is affected primarily by temperature, DIC levels and A
directly
with the atmosphere is affected primarily by temperature, DIC levels and A![]() .
While the water temperature is regulated by physical processes, including solar
energy input, sea-air heat exchanges and mixed-layer thickness, the DIC and
A
.
While the water temperature is regulated by physical processes, including solar
energy input, sea-air heat exchanges and mixed-layer thickness, the DIC and
A![]() are primarily
controlled by the biological processes of photosynthesis and respiration and
by upwelling of subsurface waters rich in respired CO
are primarily
controlled by the biological processes of photosynthesis and respiration and
by upwelling of subsurface waters rich in respired CO![]() and
nutrients. In a parcel of seawater with constant chemical composition, pCO
and
nutrients. In a parcel of seawater with constant chemical composition, pCO![]() would
increase by a factor of 4 when the water is warmed from polar temperatures
of about –1.9°C to equatorial temperatures of about 30°C. On
the other hand, the DIC in the surface ocean varies from an average value of
2150 µmol kg
would
increase by a factor of 4 when the water is warmed from polar temperatures
of about –1.9°C to equatorial temperatures of about 30°C. On
the other hand, the DIC in the surface ocean varies from an average value of
2150 µmol kg![]() in
polar regions to 1850 µmol kg
in
polar regions to 1850 µmol kg![]() in
the tropics as a result of biological processes. This change should reduce pCO
in
the tropics as a result of biological processes. This change should reduce pCO![]() by
a factor of 4. On a global scale, therefore, the magnitude of the effect of
biological drawdown on surface water pCO
by
a factor of 4. On a global scale, therefore, the magnitude of the effect of
biological drawdown on surface water pCO![]() is
similar in magnitude to the effect of temperature, but the two effects are
often compensating. Accordingly, the distribution of pCO
is
similar in magnitude to the effect of temperature, but the two effects are
often compensating. Accordingly, the distribution of pCO![]() in
surface waters in space and time, and therefore the oceanic uptake and release
of CO
in
surface waters in space and time, and therefore the oceanic uptake and release
of CO![]() , is governed
by a balance between the changes in seawater temperature, net biological utilization
of CO
, is governed
by a balance between the changes in seawater temperature, net biological utilization
of CO![]() and the
upwelling flux of subsurface waters rich in CO
and the
upwelling flux of subsurface waters rich in CO![]() .
.
Surface-water pCO![]() has
been determined with a high precision (±2 µatm) using underway
equilibrator-CO
has
been determined with a high precision (±2 µatm) using underway
equilibrator-CO![]() analyzer
systems over the global ocean since the International Geophysical Year of 1956–59.
As a result of recent major oceanographic programs, including the global CO
analyzer
systems over the global ocean since the International Geophysical Year of 1956–59.
As a result of recent major oceanographic programs, including the global CO![]() survey
and other international field studies, the database for surface-water pCO
survey
and other international field studies, the database for surface-water pCO![]() observations
has been improved to about 1 million measurements with several million accompanying
measurements of SST, salinity and other necessary parameters such as barometric
pressure and atmospheric CO
observations
has been improved to about 1 million measurements with several million accompanying
measurements of SST, salinity and other necessary parameters such as barometric
pressure and atmospheric CO![]() concentrations.
Based upon these observations, a global, monthly climatological distribution
of surface-water pCO
concentrations.
Based upon these observations, a global, monthly climatological distribution
of surface-water pCO![]() in
the ocean was created for a reference year 1995, chosen because it was the
median year of pCO
in
the ocean was created for a reference year 1995, chosen because it was the
median year of pCO![]() observations
in the database. The database and the computational method used for interpolation
of the data in space and time will be briefly described below.
observations
in the database. The database and the computational method used for interpolation
of the data in space and time will be briefly described below.
For the construction of climatological distribution maps, observations made
in different years need to be corrected to a single reference year (1995),
based on several assumptions explained below (see also Takahashi
et al., 2002). Surface waters in the subtropical gyres mix vertically
at slow rates with subsurface waters because of strong stratification at the
base of the mixed layer. As a result, they are in contact with the atmosphere
and can exchange CO![]() for
a long time. Consequently, the pCO
for
a long time. Consequently, the pCO![]() in
these warm waters follows the increasing trend of atmospheric CO
in
these warm waters follows the increasing trend of atmospheric CO![]() concentrations,
as observed by Inoue
et al. (1995) in the western North Pacific, by Feely
et al. (1999) in the
equatorial Pacific and by Bates
(2001) near Bermuda in the western North Atlantic.
Accordingly, the pCO
concentrations,
as observed by Inoue
et al. (1995) in the western North Pacific, by Feely
et al. (1999) in the
equatorial Pacific and by Bates
(2001) near Bermuda in the western North Atlantic.
Accordingly, the pCO![]() measured
in a given month and year is corrected to the same month in the reference year
1995 using the following atmospheric CO
measured
in a given month and year is corrected to the same month in the reference year
1995 using the following atmospheric CO![]() concentration
data for the planetary boundary layer: the GLOBALVIEW-CO2 database
(2000) for observations made after 1979 and the Mauna Loa data of Keeling
and Whorf (2000) for observations before 1979 (reported in CDIAC NDP-001,
revision 7).
concentration
data for the planetary boundary layer: the GLOBALVIEW-CO2 database
(2000) for observations made after 1979 and the Mauna Loa data of Keeling
and Whorf (2000) for observations before 1979 (reported in CDIAC NDP-001,
revision 7).
In contrast to the waters of the subtropical gyres, surface waters in high-latitude
regions are mixed convectively with deep waters during fall and winter, and
their CO![]() properties
tend to remain unchanged from year to year. They reflect those of the deep
waters, in which the effect of increased atmospheric CO
properties
tend to remain unchanged from year to year. They reflect those of the deep
waters, in which the effect of increased atmospheric CO![]() over
the time span of the observations is diluted to undetectable levels (Takahashi
et al., 2002). Thus no correction is necessary for the year of measurements.
over
the time span of the observations is diluted to undetectable levels (Takahashi
et al., 2002). Thus no correction is necessary for the year of measurements.
Figure 3 shows the distribution of climatological
mean sea-air pCO![]() difference
(
difference
(![]() pCO
pCO![]() )
during February (Figure 3a) and August (Figure
3b) for the reference year 1995. The yellow-red colors indicate oceanic
areas where there is a net release of CO
)
during February (Figure 3a) and August (Figure
3b) for the reference year 1995. The yellow-red colors indicate oceanic
areas where there is a net release of CO![]() to
the atmosphere, and the blue-purple colors indicate regions where there is
a net uptake of CO
to
the atmosphere, and the blue-purple colors indicate regions where there is
a net uptake of CO![]() .
The equatorial Pacific is a strong source of CO
.
The equatorial Pacific is a strong source of CO![]() to
the atmosphere throughout the year as a result of the upwelling and vertical
mixing of deep waters in the central and eastern regions of the equatorial
zone. The intensity of the oceanic release of CO
to
the atmosphere throughout the year as a result of the upwelling and vertical
mixing of deep waters in the central and eastern regions of the equatorial
zone. The intensity of the oceanic release of CO![]() decreases
westward in spite of warmer temperatures to the west. High levels of CO
decreases
westward in spite of warmer temperatures to the west. High levels of CO![]() are
released in parts of the northwestern subarctic Pacific during the northern
winter and the Arabian Sea in the Indian Ocean during August. Strong convective
mixing that brings up deep waters rich in CO
are
released in parts of the northwestern subarctic Pacific during the northern
winter and the Arabian Sea in the Indian Ocean during August. Strong convective
mixing that brings up deep waters rich in CO![]() produces
the net release of CO
produces
the net release of CO![]() in
the subarctic Pacific. The effect of increased DIC concentration surpasses
the cooling effect on pCO
in
the subarctic Pacific. The effect of increased DIC concentration surpasses
the cooling effect on pCO![]() in
seawater during winter. The high pCO
in
seawater during winter. The high pCO![]() in
the Arabian Sea water is a result of strong upwelling in response to the southwest
monsoon. High pCO
in
the Arabian Sea water is a result of strong upwelling in response to the southwest
monsoon. High pCO![]() values
in these areas are reduced by the intense primary production that follows the
periods of upwelling.
values
in these areas are reduced by the intense primary production that follows the
periods of upwelling.
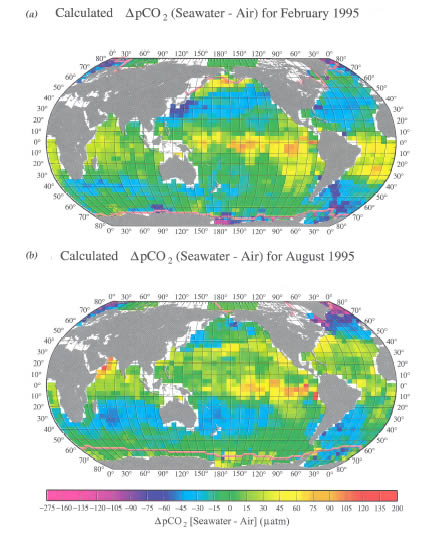
Figure 3. Distribution of climatological mean sea-air pCO![]() difference
(
difference
(![]() pCO
pCO![]() )
for the reference year 1995 representing non-El Niño conditions in February
(a) and August (b). These maps are based on about 940,000 measurements of
surface water pCO
)
for the reference year 1995 representing non-El Niño conditions in February
(a) and August (b). These maps are based on about 940,000 measurements of
surface water pCO![]() from
1958 through 2000. The pink lines indicate the edges of ice fields. The yellow-red
colors indicate regions with a net release of CO
from
1958 through 2000. The pink lines indicate the edges of ice fields. The yellow-red
colors indicate regions with a net release of CO![]() into
the atmosphere, and the blue-purple colors indicate regions with a net uptake
of CO
into
the atmosphere, and the blue-purple colors indicate regions with a net uptake
of CO![]() from
the atmosphere. The mean monthly atmospheric pCO
from
the atmosphere. The mean monthly atmospheric pCO![]() value
in each pixel in 1995, (pCO
value
in each pixel in 1995, (pCO![]() )air,
is computed using (pCO
)air,
is computed using (pCO![]() )air
= (CO
)air
= (CO![]() )air × (Pb
- pH2O). (CO
)air × (Pb
- pH2O). (CO![]() )air
is the monthly mean atmospheric CO
)air
is the monthly mean atmospheric CO![]() concentration
(mole fraction of CO
concentration
(mole fraction of CO![]() in
dry air) from the GLOBALVIEW
database (2000); Pb is the climatological mean barometric pressure
at sea level from the Atlas
of Surface Marine Data (1994); and the water vapor pressure, pH
in
dry air) from the GLOBALVIEW
database (2000); Pb is the climatological mean barometric pressure
at sea level from the Atlas
of Surface Marine Data (1994); and the water vapor pressure, pH![]() O,
is computed using the mixed layer water temperature and salinity from the
World Ocean Database (1998) of NODC/NOAA. The sea-air pCO
O,
is computed using the mixed layer water temperature and salinity from the
World Ocean Database (1998) of NODC/NOAA. The sea-air pCO![]() difference
values in the reference year 1995 have been computed by subtracting the mean
monthly atmospheric pCO
difference
values in the reference year 1995 have been computed by subtracting the mean
monthly atmospheric pCO![]() value
from the mean monthly surface ocean water pCO
value
from the mean monthly surface ocean water pCO![]() value
in each pixel.
value
in each pixel.
The temperate regions of the North Pacific and Atlantic oceans take up a moderate
amount of CO![]() (blue)
during the northern winter (Figure 3a) and release
a moderate amount (yellow-green) during the northern summer (Figure
3b). This pattern is the result primarily of seasonal temperature changes.
Similar seasonal changes are observed in the southern temperate oceans. Intense
regions of CO
(blue)
during the northern winter (Figure 3a) and release
a moderate amount (yellow-green) during the northern summer (Figure
3b). This pattern is the result primarily of seasonal temperature changes.
Similar seasonal changes are observed in the southern temperate oceans. Intense
regions of CO![]() uptake
(blue-purple) are seen in the high-latitude northern ocean in summer (Figure
3b) and in the high-latitude South Atlantic and Southern oceans near Antarctica
in austral summer (Figure 3a). The uptake is
linked to high biological utilization of CO
uptake
(blue-purple) are seen in the high-latitude northern ocean in summer (Figure
3b) and in the high-latitude South Atlantic and Southern oceans near Antarctica
in austral summer (Figure 3a). The uptake is
linked to high biological utilization of CO![]() in
thin mixed layers. As the seasons progress, vertical mixing of deep waters
eliminates the uptake of CO
in
thin mixed layers. As the seasons progress, vertical mixing of deep waters
eliminates the uptake of CO![]() .
.
These observations point out that the ![]() pCO
pCO![]() in
high-latitude oceans is governed primarily by deepwater upwelling in winter
and biological uptake in spring and summer, whereas in the temperate and subtropical
oceans, the
in
high-latitude oceans is governed primarily by deepwater upwelling in winter
and biological uptake in spring and summer, whereas in the temperate and subtropical
oceans, the ![]() pCO
pCO![]() is
governed primarily by water temperature. The seawater
is
governed primarily by water temperature. The seawater ![]() pCO
pCO![]() is
highest during winter in subpolar and polar waters, whereas it is highest during
summer in the temperate regions. Thus the seasonal variation of
is
highest during winter in subpolar and polar waters, whereas it is highest during
summer in the temperate regions. Thus the seasonal variation of ![]() pCO
pCO![]() and
therefore the shift between net uptake and release of CO
and
therefore the shift between net uptake and release of CO![]() in
subpolar and polar regions is about 6 months out of phase with that in the
temperate regions.
in
subpolar and polar regions is about 6 months out of phase with that in the
temperate regions.
The ![]() pCO
pCO![]() maps
are combined with the solubility (s) in seawater and the kinetic forcing function,
the gas transfer velocity (k), to produce the flux:
maps
are combined with the solubility (s) in seawater and the kinetic forcing function,
the gas transfer velocity (k), to produce the flux:
F = k•s•![]() pCO
pCO![]() (1)
(1)
The gas transfer velocity is controlled by near-surface turbulence in the liquid boundary layer. Laboratory studies in wind-wave tanks have shown that k is a strong but non-unique function of wind speed. The results from various wind-wave tank investigations and field studies indicate that factors such as fetch, wave direction, atmospheric boundary layer stability and bubble entrainment influence the rate of gas transfer. Also, surfactants can inhibit gas exchange through their damping effect on waves. Since effects other than wind speed have not been well quantified, the processes controlling gas transfer have been parameterized solely with wind speed, in large part because k is strongly dependent on wind, and global and regional wind-speed data are readily available.
Several of the frequently used relationships for the estimation of gas transfer velocity as a function of wind speed are shown in Figure 4 to illustrate their different dependencies. For the Liss and Merlivat (1986) relationship, the slope and intercept of the lower segment was determined from an analytical solution of transfer across a smooth boundary. For the intermediate wind regime, the middle segment was obtained from a field study in a small lake, and results from a wind-wave tank study were used for the high wind regime after applying some adjustments. This relationship is often considered the lower bound of gas transfer-wind speed relationships.
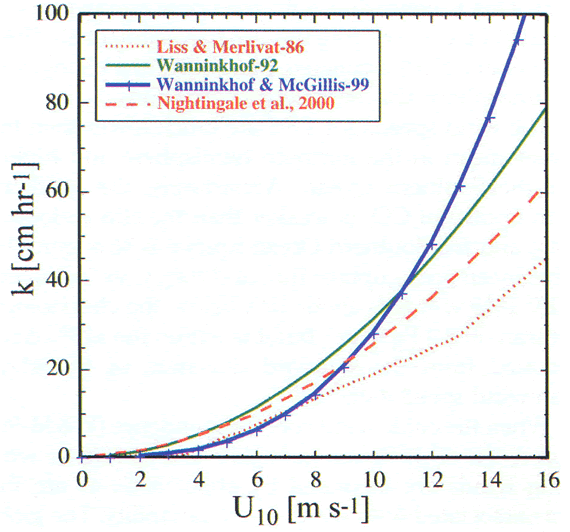
Figure 4. Graph of the different relationships that have been developed
for the estimation of the gas transfer velocity, k, as a function
of wind speed. The relationships were developed from wind-wave tank experiments,
oceanic observations, global constraints and basic theory. The different
forms of the relationships are summarized in Table
1. U![]() is
wind speed at 10 m above the sea surface.
is
wind speed at 10 m above the sea surface.
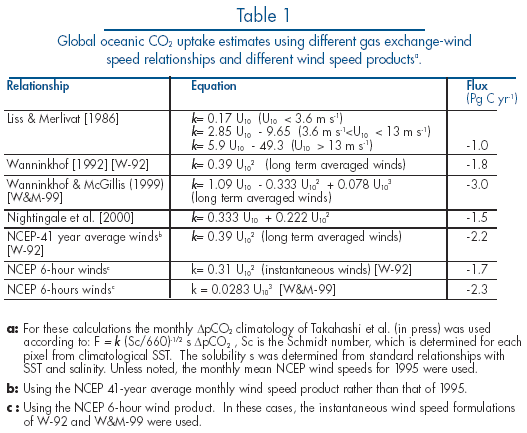
The quadratic relationship of Wanninkhof
(1992) was constructed to follow the general shape of curves derived
in wind-wave tanks but adjusted so that the global mean transfer velocity
corresponds with the long-term global average gas transfer velocity determined
from the invasion of bomb ![]() C
into the ocean. Because the bomb
C
into the ocean. Because the bomb ![]() C
is also used as a diagnostic or tuning parameter in global ocean biogeochemical
circulation models, this parameterization yields internally consistent results
when used with these models, making it one of the more favored parameterizations.
C
is also used as a diagnostic or tuning parameter in global ocean biogeochemical
circulation models, this parameterization yields internally consistent results
when used with these models, making it one of the more favored parameterizations.
Using the same long-term global ![]() C
constraint but basing the general shape of the curve on recent CO
C
constraint but basing the general shape of the curve on recent CO![]() flux
observations over the North Atlantic determined using the covariance technique, Wanninkhof
and McGillis (1999) proposed a significantly stronger (cubic) dependence
with wind speed. This relationship shows a weaker dependence on wind for wind
speeds less than 10 ms
flux
observations over the North Atlantic determined using the covariance technique, Wanninkhof
and McGillis (1999) proposed a significantly stronger (cubic) dependence
with wind speed. This relationship shows a weaker dependence on wind for wind
speeds less than 10 ms![]() and
a significantly stronger dependence at higher wind speeds. However, the relationship
is not well constrained at high wind speeds because of the large scatter in
the scarce observations. Both the U
and
a significantly stronger dependence at higher wind speeds. However, the relationship
is not well constrained at high wind speeds because of the large scatter in
the scarce observations. Both the U![]() and
U
and
U![]() relationships
fit within the data envelope of the study, but the U
relationships
fit within the data envelope of the study, but the U![]() relationship
provides a significantly better fit. Nightingale
et al. (2000) determined a gas exchange-wind speed relationship based on
the results of a series of experiments utilizing deliberately injected sulfur
hexafluoride (SF
relationship
provides a significantly better fit. Nightingale
et al. (2000) determined a gas exchange-wind speed relationship based on
the results of a series of experiments utilizing deliberately injected sulfur
hexafluoride (SF![]() ),
), ![]() He
and non-volatile tracers performed in the last decade.
He
and non-volatile tracers performed in the last decade.
The global oceanic CO![]() uptake
using different wind speed/gas transfer velocity parameterizations differs
by a factor of three (Table 1). The wide range
of global CO
uptake
using different wind speed/gas transfer velocity parameterizations differs
by a factor of three (Table 1). The wide range
of global CO![]() fluxes
for the different relationships illustrates the large range of results and
assumptions that are used to produce these relationships. Aside from differences
in global oceanic CO
fluxes
for the different relationships illustrates the large range of results and
assumptions that are used to produce these relationships. Aside from differences
in global oceanic CO![]() uptake,
there are also significant regional differences. Figure
5 shows that the relationship of W&M-99 yields systematically lower
evasion rates in the equatorial region and higher uptake rates at high latitudes
compared with W-92, leading to significantly larger global CO
uptake,
there are also significant regional differences. Figure
5 shows that the relationship of W&M-99 yields systematically lower
evasion rates in the equatorial region and higher uptake rates at high latitudes
compared with W-92, leading to significantly larger global CO![]() uptake
estimates.
uptake
estimates.
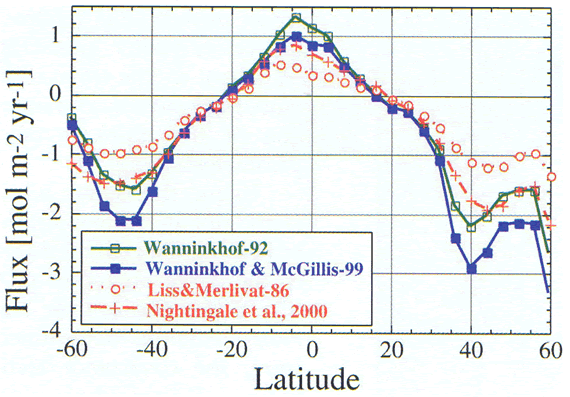
Figure 5. Effects of the various gas transfer/wind speed relationships
on the estimated air-sea exchange flux of CO![]() in
the ocean as a function of latitude. The global effects on the net air-sea
flux are given in Table 1.
in
the ocean as a function of latitude. The global effects on the net air-sea
flux are given in Table 1.
In addition to the non-unique dependence of gas exchange on wind speed, which
causes a large spread in global air-sea CO![]() flux
estimates, there are several other factors contributing to biases in the results.
Global wind-speed data obtained from shipboard observations, satellites and
data assimilation techniques show significant differences on regional and global
scales. Because of the non-linearity of the relationships between gas exchange
and wind speed, significant biases are introduced in methods of averaging the
product of gas transfer velocity and wind speed. The common approach of averaging
the
flux
estimates, there are several other factors contributing to biases in the results.
Global wind-speed data obtained from shipboard observations, satellites and
data assimilation techniques show significant differences on regional and global
scales. Because of the non-linearity of the relationships between gas exchange
and wind speed, significant biases are introduced in methods of averaging the
product of gas transfer velocity and wind speed. The common approach of averaging
the ![]() pCO
pCO![]() and k separately
over monthly periods, determining the flux from the product and ignoring the
cross product leads to a bias that is about 0.2 to 0.8 Pg C yr
and k separately
over monthly periods, determining the flux from the product and ignoring the
cross product leads to a bias that is about 0.2 to 0.8 Pg C yr![]() lower
in the global uptake estimate. This bias shows a regional variation that is
dependent on the distribution and magnitude of winds. This issue has been partly
rectified in some of the relationships in which a global wind-speed distribution
is used to create separate relationships between gas transfer and wind speed
for short-term (a day or less) and long-term (a month or more) periods. Since
wind-speed distributions are regionally dependent and vary on time scales of
hours, this approach is far from perfect.
lower
in the global uptake estimate. This bias shows a regional variation that is
dependent on the distribution and magnitude of winds. This issue has been partly
rectified in some of the relationships in which a global wind-speed distribution
is used to create separate relationships between gas transfer and wind speed
for short-term (a day or less) and long-term (a month or more) periods. Since
wind-speed distributions are regionally dependent and vary on time scales of
hours, this approach is far from perfect.
The groundwork of efforts laid over the past decade and recently improved
technologies make the quantification of regional and global CO![]() fluxes
a more tractable problem now. Satellites equipped with scatterometers that
are used to determine wind speed offer daily global coverage. Moreover, these
instruments measure sea-surface roughness that is directly related to gas transfer.
This remotely sensed information, along with regional statistics of wind-speed
variability on time scales shorter than a day, offers the real possibility
that more accurate gas transfer velocities will be obtained. Efforts are underway
to increase the coverage of pCO
fluxes
a more tractable problem now. Satellites equipped with scatterometers that
are used to determine wind speed offer daily global coverage. Moreover, these
instruments measure sea-surface roughness that is directly related to gas transfer.
This remotely sensed information, along with regional statistics of wind-speed
variability on time scales shorter than a day, offers the real possibility
that more accurate gas transfer velocities will be obtained. Efforts are underway
to increase the coverage of pCO![]() through
more frequent measurements and data assimilation techniques, again utilizing
remote sensing of parameters such as sea-surface temperature and wind speed.
Better quantification of the fluxes will lead to better boundary conditions
for models and improved forecasts of atmospheric CO
through
more frequent measurements and data assimilation techniques, again utilizing
remote sensing of parameters such as sea-surface temperature and wind speed.
Better quantification of the fluxes will lead to better boundary conditions
for models and improved forecasts of atmospheric CO![]() concentrations.
concentrations.
To illustrate the sensitivity of the gas transfer velocity and thus the sea-air
CO![]() flux to wind
speed, we have estimated the regional and global net sea-air CO
flux to wind
speed, we have estimated the regional and global net sea-air CO![]() fluxes
using two different formulations for the CO
fluxes
using two different formulations for the CO![]() gas
transfer coefficient across the sea-air interface: the quadratic U
gas
transfer coefficient across the sea-air interface: the quadratic U![]() dependence
of W-92 and the cubic U
dependence
of W-92 and the cubic U![]() dependence
of W&M-99. In addition, we have demonstrated the effects of wind-speed
fields on the computed sea-air CO
dependence
of W&M-99. In addition, we have demonstrated the effects of wind-speed
fields on the computed sea-air CO![]() flux
using the National Center for Environmental Prediction (NCEP)-41 mean monthly
wind speed and the NCEP-1995 mean monthly wind speed distributions over 4° × 5° pixel
areas.
flux
using the National Center for Environmental Prediction (NCEP)-41 mean monthly
wind speed and the NCEP-1995 mean monthly wind speed distributions over 4° × 5° pixel
areas.
In Table 2 the fluxes computed using the
W-92 and the NCEP/National Center for Atmospheric Research (NCAR) 41-year mean
wind are listed in the first row for each grouping in column one (for latitudinal
bands, oceanic regions and regional flux). The column "Errors in Flux" located
at the extreme right of Table 2 lists the
deviations from the mean flux that have been determined by adding or subtracting
one standard deviation of the wind speed (about ±2 m sec![]() on
the global average) from the mean monthly wind speed in each pixel area. These
changes in wind speeds affect the regional and global flux values by about ±25%.
The fluxes computed using the single year mean wind speed data for 1995 are
listed in the second line in each column one grouping in the table.
on
the global average) from the mean monthly wind speed in each pixel area. These
changes in wind speeds affect the regional and global flux values by about ±25%.
The fluxes computed using the single year mean wind speed data for 1995 are
listed in the second line in each column one grouping in the table.
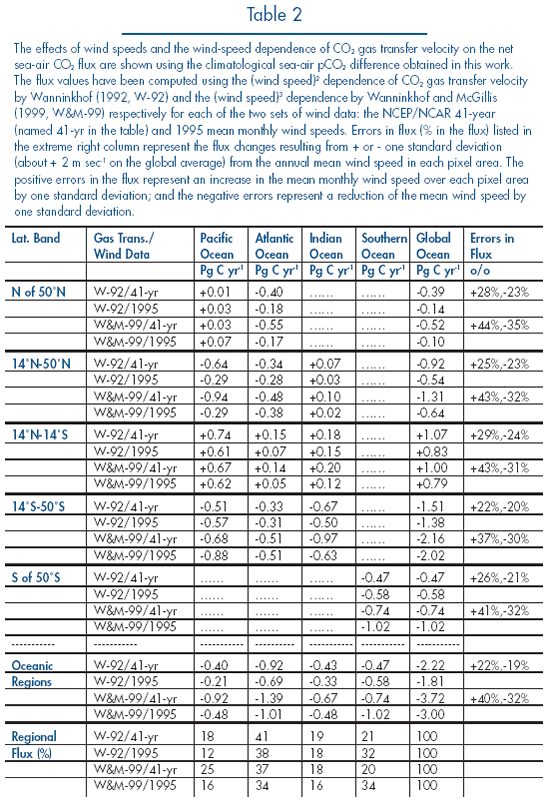
The global ocean uptake estimated using the W-92 and the NCEP 41-yr mean wind
speeds is –2.2 ± 0.4 Pg C yr![]() .
This is consistent with the ocean uptake flux of –2.0 ± 0.6 Pg
C yr
.
This is consistent with the ocean uptake flux of –2.0 ± 0.6 Pg
C yr![]() during
the 1990s (Keeling
et al., 1996; Battle
et al., 2000) estimated from observed changes in the atmospheric CO
during
the 1990s (Keeling
et al., 1996; Battle
et al., 2000) estimated from observed changes in the atmospheric CO![]() and
oxygen variations.
and
oxygen variations.
The wind speeds for 1995 are much lower than the 41-year mean in the northern
hemisphere and higher over the Southern Ocean. Accordingly, the northern ocean
uptake of CO![]() is
weaker than the climatological mean, and the Southern Ocean uptake is stronger.
The global mean ocean uptake flux of –1.8 Pg C yr
is
weaker than the climatological mean, and the Southern Ocean uptake is stronger.
The global mean ocean uptake flux of –1.8 Pg C yr![]() using
the NCEP-1995 winds is about 18% below the climatological mean of –2.2 Pg C
yr
using
the NCEP-1995 winds is about 18% below the climatological mean of –2.2 Pg C
yr![]() ,
but it is within the ±25% error estimated from the standard deviation
of the 41-yr mean wind speed data.
,
but it is within the ±25% error estimated from the standard deviation
of the 41-yr mean wind speed data.
When the cubic wind speed dependence (W&M-99) is used, the CO![]() fluxes
in higher latitude areas with strong winds are increased by about 50%, as are
the errors associated with wind speed variability. The global ocean uptake
flux computed with the 41-year mean wind speed data and the NCEP-1995 wind
data is –3.7 Pg C yr
fluxes
in higher latitude areas with strong winds are increased by about 50%, as are
the errors associated with wind speed variability. The global ocean uptake
flux computed with the 41-year mean wind speed data and the NCEP-1995 wind
data is –3.7 Pg C yr![]() and
–3.0 Pg C yr
and
–3.0 Pg C yr![]() respectively,
an increase of about 70% over the fluxes computed from the W-92 dependence.
These flux values are significantly greater than the flux based on atmospheric
CO
respectively,
an increase of about 70% over the fluxes computed from the W-92 dependence.
These flux values are significantly greater than the flux based on atmospheric
CO![]() and oxygen
data (Keeling
et al., 1996; Battle
et al., 2000). However, the relative magnitudes of CO
and oxygen
data (Keeling
et al., 1996; Battle
et al., 2000). However, the relative magnitudes of CO![]() uptake
by ocean basins (shown in % in the regional flux grouping in the last four
rows of Table 2) remain nearly unaffected
by the choice of the wind-speed dependence of the gas transfer velocity.
uptake
by ocean basins (shown in % in the regional flux grouping in the last four
rows of Table 2) remain nearly unaffected
by the choice of the wind-speed dependence of the gas transfer velocity.
The distribution of winds can also influence the calculated gas transfer velocity.
This is because of the nonlinear dependence of gas exchange with wind speed;
long-term average winds underestimate flux especially for strongly non-linear
dependencies. To avoid this bias, the relationships are adjusted by assuming
that the global average wind speed is well represented by a Rayleigh distribution
function. As noted by Wanninkhof
et al. (2001), this overestimates the flux. A more appropriate way
to deal with the issue of wind speed variability is to use short-term winds.
If the NCEP 6-hour wind products are used, the global flux computed using the
W&M-99 cubic wind-speed formulation decreases from –3.7 to –3.0
Pg C yr![]() for
the NCEP 41-year winds and from –3.0 to –2.3 Pg C yr
for
the NCEP 41-year winds and from –3.0 to –2.3 Pg C yr![]() for
the NCEP 1995 wind data.
for
the NCEP 1995 wind data.
The relative importance of the major ocean basins in the ocean uptake of CO![]() may
be assessed on the basis of the CO
may
be assessed on the basis of the CO![]() fluxes
obtained from our pCO
fluxes
obtained from our pCO![]() data
and W-92 gas transfer velocity (Table 2 and Figure
6). The Atlantic Ocean as a whole, which has 23.5% of the global ocean
area, is the region with the strongest net CO
data
and W-92 gas transfer velocity (Table 2 and Figure
6). The Atlantic Ocean as a whole, which has 23.5% of the global ocean
area, is the region with the strongest net CO![]() uptake
(41%). The high-latitude northern North Atlantic, including the Greenland,
Iceland and Norwegian seas, is responsible for a substantial amount of this
CO
uptake
(41%). The high-latitude northern North Atlantic, including the Greenland,
Iceland and Norwegian seas, is responsible for a substantial amount of this
CO![]() uptake while
representing only 5% of the global ocean in area. This reflects a combination
of two factors: the intense summertime primary production and the low CO
uptake while
representing only 5% of the global ocean in area. This reflects a combination
of two factors: the intense summertime primary production and the low CO![]() concentrations
in subsurface waters associated with recent ventilation of North Atlantic subsurface
waters. The Pacific Ocean as a whole takes up the smallest amount of CO
concentrations
in subsurface waters associated with recent ventilation of North Atlantic subsurface
waters. The Pacific Ocean as a whole takes up the smallest amount of CO![]() (18%
of the total) in spite of its size (49% of the total ocean area). This is because
mid-latitude uptake (about 1.1 Pg C yr
(18%
of the total) in spite of its size (49% of the total ocean area). This is because
mid-latitude uptake (about 1.1 Pg C yr![]() )
is almost compensated for by the large equatorial release of about 0.7 Pg C
yr
)
is almost compensated for by the large equatorial release of about 0.7 Pg C
yr![]() .
If the equatorial flux were totally eliminated, as during very strong El Niño
conditions, the Pacific would take up CO
.
If the equatorial flux were totally eliminated, as during very strong El Niño
conditions, the Pacific would take up CO![]() to
an extent comparable to the entire North and South Atlantic Ocean. The southern
Indian Ocean is a region of strong uptake in spite of its small area (15% of
the total). This may be attributed primarily to the cooling of tropical waters
flowing southward in the western South Indian Ocean.
to
an extent comparable to the entire North and South Atlantic Ocean. The southern
Indian Ocean is a region of strong uptake in spite of its small area (15% of
the total). This may be attributed primarily to the cooling of tropical waters
flowing southward in the western South Indian Ocean.
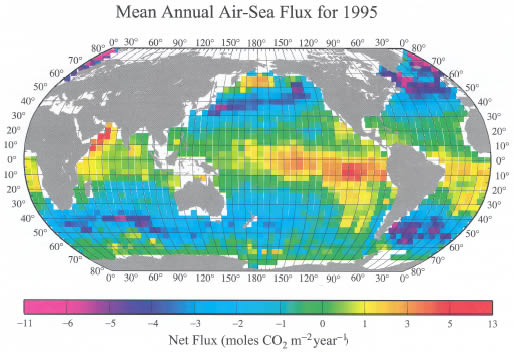
Figure 6. Distribution of the climatological mean annual sea-air CO![]() flux
(moles CO
flux
(moles CO![]() m
m![]() yr
yr![]() )
for the reference year 1995 representing non-El Niño conditions. This
has been computed using the mean monthly distribution of sea-air pCO
)
for the reference year 1995 representing non-El Niño conditions. This
has been computed using the mean monthly distribution of sea-air pCO![]() difference,
the climatological NCEP 41-year mean wind speed and the wind-speed dependence
of the CO
difference,
the climatological NCEP 41-year mean wind speed and the wind-speed dependence
of the CO![]() gas
transfer velocity of Wanninkhof
(1992). The yellow-red colors indicate a region characterized by a net
release of CO
gas
transfer velocity of Wanninkhof
(1992). The yellow-red colors indicate a region characterized by a net
release of CO![]() to
the atmosphere, and the blue-purple colors indicate a region with a net uptake
of CO
to
the atmosphere, and the blue-purple colors indicate a region with a net uptake
of CO![]() from
the atmosphere. This map yields an annual oceanic uptake flux for CO
from
the atmosphere. This map yields an annual oceanic uptake flux for CO![]() of
2.2 ± 0.4 Pg C yr
of
2.2 ± 0.4 Pg C yr![]() .
.
To understand the role of the oceans as a sink for anthropogenic CO![]() ,
it is important to determine the distribution of carbon species in the ocean
interior and the processes affecting the transport and storage of CO
,
it is important to determine the distribution of carbon species in the ocean
interior and the processes affecting the transport and storage of CO![]() taken
up from the atmosphere. Figure 7 shows the typical
north-south distribution of DIC in the Atlantic, Indian, and Pacific oceans
prior to the introduction of anthropogenic CO
taken
up from the atmosphere. Figure 7 shows the typical
north-south distribution of DIC in the Atlantic, Indian, and Pacific oceans
prior to the introduction of anthropogenic CO![]() .
In general, DIC is about 10–15% higher in deep waters than at the surface.
Concentrations are also generally lower in the Atlantic than the Indian ocean,
with the highest concentrations found in the older deep waters of the North
Pacific. The two basic mechanisms that control the distribution of carbon in
the oceans are the solubility and biological pumps.
.
In general, DIC is about 10–15% higher in deep waters than at the surface.
Concentrations are also generally lower in the Atlantic than the Indian ocean,
with the highest concentrations found in the older deep waters of the North
Pacific. The two basic mechanisms that control the distribution of carbon in
the oceans are the solubility and biological pumps.
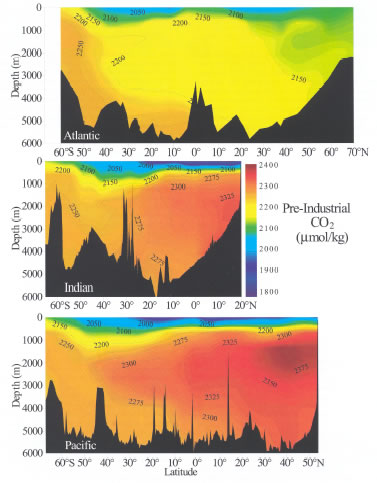
Figure 7. Zonal mean pre-industrial distributions of dissolved inorganic
carbon (in units of µmol kg![]() )
along north-south transects in the Atlantic, Indian and Pacific oceans. The
Pacific and Indian Ocean data are from the Global CO
)
along north-south transects in the Atlantic, Indian and Pacific oceans. The
Pacific and Indian Ocean data are from the Global CO![]() Survey
(this study), and the Atlantic Ocean data are from Gruber
(1998).
Survey
(this study), and the Atlantic Ocean data are from Gruber
(1998).
The solubility pump is driven by two interrelated factors. First, CO![]() is
more than twice as soluble in cold polar waters than in warm equatorial waters.
As western surface boundary currents transport water from the tropics to the
poles, the waters are cooled and absorb more CO
is
more than twice as soluble in cold polar waters than in warm equatorial waters.
As western surface boundary currents transport water from the tropics to the
poles, the waters are cooled and absorb more CO![]() from
the atmosphere. Second, the high-latitude zones are also regions where intermediate
and bottom waters are formed. As these waters cool, they become denser and
sink into the ocean interior, taking with them the CO
from
the atmosphere. Second, the high-latitude zones are also regions where intermediate
and bottom waters are formed. As these waters cool, they become denser and
sink into the ocean interior, taking with them the CO![]() accumulated
at the surface.
accumulated
at the surface.
The primary production of marine phytoplankton transforms CO![]() and
nutrients from seawater into organic material. Although most of the CO
and
nutrients from seawater into organic material. Although most of the CO![]() taken
up by phytoplankton is recycled near the surface, a substantial fraction, perhaps
30%, sinks into the deeper waters before being converted back into CO
taken
up by phytoplankton is recycled near the surface, a substantial fraction, perhaps
30%, sinks into the deeper waters before being converted back into CO![]() by
marine bacteria. Only about 0.1% reaches the seafloor to be buried in the sediments.
The CO
by
marine bacteria. Only about 0.1% reaches the seafloor to be buried in the sediments.
The CO![]() that
is recycled at depth is slowly transported over long distances by the largescale
thermohaline circulation. DIC slowly accumulates in the deep waters as they
travel from the Atlantic to the Indian and Pacific oceans. Using a 3-D global
carbon model, Sarmiento
et al. (1995) estimated that the natural solubility
pump is responsible for about 20% of the vertical gradient in DIC; the remaining
80% originates from the biological pump.
that
is recycled at depth is slowly transported over long distances by the largescale
thermohaline circulation. DIC slowly accumulates in the deep waters as they
travel from the Atlantic to the Indian and Pacific oceans. Using a 3-D global
carbon model, Sarmiento
et al. (1995) estimated that the natural solubility
pump is responsible for about 20% of the vertical gradient in DIC; the remaining
80% originates from the biological pump.
The approaches for estimating anthropogenic CO![]() in
the oceans have taken many turns over the past decade. Siegenthaler
and Sarmiento (1993) summarized early approaches for estimating the anthropogenic
sink in the oceans, including ocean models of various complexity, atmospheric
measurements and transport models used together with pCO
in
the oceans have taken many turns over the past decade. Siegenthaler
and Sarmiento (1993) summarized early approaches for estimating the anthropogenic
sink in the oceans, including ocean models of various complexity, atmospheric
measurements and transport models used together with pCO![]() measurements
and estimates based on changes in oceanic
measurements
and estimates based on changes in oceanic ![]() C
and oxygen mass balance. They noted the wide range of ocean uptake estimates
(1.6–2.3 Pg C yr
C
and oxygen mass balance. They noted the wide range of ocean uptake estimates
(1.6–2.3 Pg C yr![]() )
and concluded that the larger uptake estimates from the models were the most
reliable.
)
and concluded that the larger uptake estimates from the models were the most
reliable.
The first approaches for using measurements to isolate anthropogenic CO![]() from
the large, natural DIC signal were independently proposed by Brewer
(1978) and Chen
and Millero (1979). Both these approaches were based on the premise that
the anthropogenic DIC concentration could be isolated from the measured DIC
by subtracting the contributions of the biological pump and the physical processes,
including the pre-industrial source water values and the solubility pump.
from
the large, natural DIC signal were independently proposed by Brewer
(1978) and Chen
and Millero (1979). Both these approaches were based on the premise that
the anthropogenic DIC concentration could be isolated from the measured DIC
by subtracting the contributions of the biological pump and the physical processes,
including the pre-industrial source water values and the solubility pump.
Gruber
et al. (1996) improved the earlier approaches by developing the ![]() C*
method. This method is based on the premise that the anthropogenic CO
C*
method. This method is based on the premise that the anthropogenic CO![]() concentration
(Cant) can be isolated from measured DIC values (Cm)
by subtracting the contribution of the biological pumps (
concentration
(Cant) can be isolated from measured DIC values (Cm)
by subtracting the contribution of the biological pumps (![]() Cbio),
the DIC the waters would have in equilibrium with a preindustrial atmospheric
CO
Cbio),
the DIC the waters would have in equilibrium with a preindustrial atmospheric
CO![]() concentration
of 280 ppm (Ceq280), and a term that corrects for the fact that
surface waters are not always in equilibrium with the atmosphere (
concentration
of 280 ppm (Ceq280), and a term that corrects for the fact that
surface waters are not always in equilibrium with the atmosphere (![]() Cdiseq):
Cdiseq):
Cant = Cm – ![]() Cbio – Ceq280 – Cdiseq =
Cbio – Ceq280 – Cdiseq = ![]() C* –
C* – ![]() Cdiseq. (2)
Cdiseq. (2)
The three terms to the right of the first equal sign make up ![]() C*,
which can be explicitly calculated for each sample. The fact that
C*,
which can be explicitly calculated for each sample. The fact that ![]() C*
is a quasi-conservative tracer helps remedy some of the mixing concerns arising
from the earlier techniques (Sabine
and Feely, 2001). The
C*
is a quasi-conservative tracer helps remedy some of the mixing concerns arising
from the earlier techniques (Sabine
and Feely, 2001). The ![]() Cdiseq term
is evaluated over small isopycnal intervals using a water-mass age tracer such
as CFCs.
Cdiseq term
is evaluated over small isopycnal intervals using a water-mass age tracer such
as CFCs.
We have evaluated anthropogenic CO![]() for
the Atlantic, Indian, and Pacific oceans using the
for
the Atlantic, Indian, and Pacific oceans using the ![]() C*
approach. Figure 8 shows representative sections
of anthropogenic CO
C*
approach. Figure 8 shows representative sections
of anthropogenic CO![]() for
each of the ocean basins. Surface values range from about 45 to 60 µmol
kg
for
each of the ocean basins. Surface values range from about 45 to 60 µmol
kg![]() .
The deepest penetrations are observed in areas of deep water formation, such
as the North Atlantic, and intermediate water formation, such as 40–50°S.
Integrated water column inventories of anthropogenic CO
.
The deepest penetrations are observed in areas of deep water formation, such
as the North Atlantic, and intermediate water formation, such as 40–50°S.
Integrated water column inventories of anthropogenic CO![]() exceed
60 moles m
exceed
60 moles m![]() in
the North Atlantic (Figure 9). Areas where older
waters are upwelled, like the high-latitude waters around Antarctica and Equatorial
Pacific waters, show relatively shallow penetration. Consequently, anthropogenic
CO
in
the North Atlantic (Figure 9). Areas where older
waters are upwelled, like the high-latitude waters around Antarctica and Equatorial
Pacific waters, show relatively shallow penetration. Consequently, anthropogenic
CO![]() inventories
are all less than 40 moles m
inventories
are all less than 40 moles m![]() in
these regions (Figure 9).
in
these regions (Figure 9).
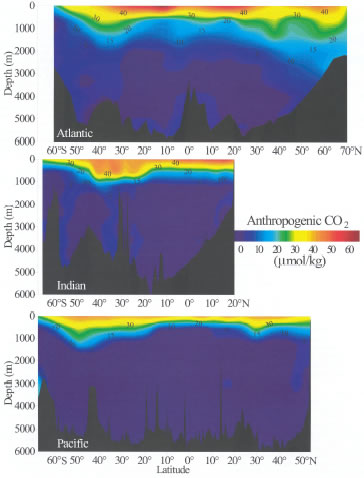
Figure 8. Zonal mean distributions of estimated anthropogenic CO![]() concentrations
(in units of µmol kg
concentrations
(in units of µmol kg![]() )
along north-south transects in the Atlantic, Indian and Pacific oceans. The
Pacific and Indian Ocean data are from the Global CO
)
along north-south transects in the Atlantic, Indian and Pacific oceans. The
Pacific and Indian Ocean data are from the Global CO![]() Survey
(this study), and the Atlantic Ocean data are from Gruber
(1998).
Survey
(this study), and the Atlantic Ocean data are from Gruber
(1998).
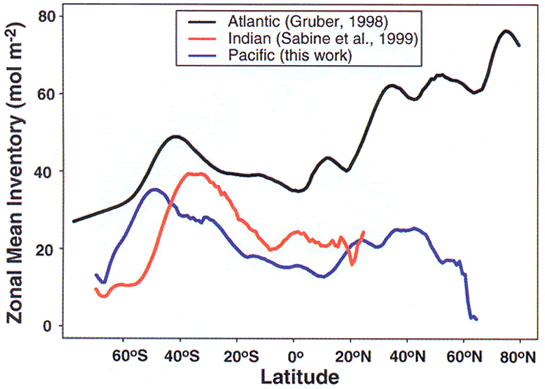
Figure 9. Zonal mean anthropogenic CO![]() inventories
(in units of moles m
inventories
(in units of moles m![]() )
in the Atlantic, Indian and Pacific oceans.
)
in the Atlantic, Indian and Pacific oceans.
Data-based estimates indicate that the oceans have taken up approximately 105 ± 8 Pg C since the beginning of the industrial era. Current global carbon models generally agree with the total inventory estimates, but discrepancies still exist in the regional distribution of the anthropogenic inventories. Some of these discrepancies stem from deficiencies in the modeled circulation and water mass formation. There are also a number of assumptions in the data-based approaches regarding the use of constant stoichiometric ratios and time-invariant air-sea disequilibria that may be inadequate in some regions. These are all areas of current research. Anthropogenic estimates should continue to converge as both the models and the data-based approaches are improved with time.
As CO![]() continues
to increase in the atmosphere, it is important to continue the work begun with
the Global Survey of CO
continues
to increase in the atmosphere, it is important to continue the work begun with
the Global Survey of CO![]() in
the Ocean. Because CO
in
the Ocean. Because CO![]() is
an acid gas, the uptake of anthropogenic CO
is
an acid gas, the uptake of anthropogenic CO![]() consumes
carbonate ions and lowers the pH of the ocean. The carbonate ion concentration
of surface seawater in equilibrium with the atmosphere will decrease by about
30% and the hydrogen ion concentration will increase by about 70% with a doubling
of atmospheric CO
consumes
carbonate ions and lowers the pH of the ocean. The carbonate ion concentration
of surface seawater in equilibrium with the atmosphere will decrease by about
30% and the hydrogen ion concentration will increase by about 70% with a doubling
of atmospheric CO![]() from
pre-industrial levels (280 to 560 ppm). As the carbonate ion concentration
decreases, the buffering capacity of the ocean and its ability to absorb more
CO
from
pre-industrial levels (280 to 560 ppm). As the carbonate ion concentration
decreases, the buffering capacity of the ocean and its ability to absorb more
CO![]() from the
atmosphere is diminished. Over the long term (millennial time scales) the ocean
has the potential to absorb as much as 85% of the anthropogenic CO
from the
atmosphere is diminished. Over the long term (millennial time scales) the ocean
has the potential to absorb as much as 85% of the anthropogenic CO![]() that
is released into the atmosphere. Because the lifetime of fossil fuel CO
that
is released into the atmosphere. Because the lifetime of fossil fuel CO![]() in
the atmosphere ranges from decades to centuries, mankind's reliance on fossil
fuel for heat and energy will continue to have a significant effect on the
chemistry of the earth's atmosphere and oceans and therefore on our climate
for many centuries to millennia.
in
the atmosphere ranges from decades to centuries, mankind's reliance on fossil
fuel for heat and energy will continue to have a significant effect on the
chemistry of the earth's atmosphere and oceans and therefore on our climate
for many centuries to millennia.
Plans are being formulated in several countries, including the United States,
to establish a set of repeat sections to document the increasing anthropogenic
inventories in the oceans. Most of these sections will follow the lines occupied
during the WOCE Hydrographic Programme on which JGOFS investigators made CO![]() survey
measurements. The current synthesis effort will provide an important baseline
for assessment of future changes in the carbon system. The spatially extensive
information from the repeat sections, together with the temporal records from
the time-series stations and the spatial and temporal records available from
automated surface pCO
survey
measurements. The current synthesis effort will provide an important baseline
for assessment of future changes in the carbon system. The spatially extensive
information from the repeat sections, together with the temporal records from
the time-series stations and the spatial and temporal records available from
automated surface pCO![]() measurements
on ships of opportunity, will greatly improve our understanding of the ocean
carbon system and provide better constraints on potential changes in the future.
measurements
on ships of opportunity, will greatly improve our understanding of the ocean
carbon system and provide better constraints on potential changes in the future.
The authors are grateful to the members of the CO![]() Science
Team and the JGOFS and WOCE investigators for making their data available for
this work. We thank Lisa Dilling of the National Oceanic and Atmospheric Administration
(NOAA) Office of Global Programs, Don Rice of the National Science Foundation
and Mike Riches of the Department of Energy (DOE) for their efforts in coordinating
this research. This work was supported by DOE and NOAA as a contribution to
the U.S. JGOFS Synthesis and Modeling Project (Grant No. GC99-220) and by grants
to Taro Takahashi from NSF (OPP-9506684) and NOAA (NA16GP01018). This publication
was supported by the Joint Institute for the Study of the Atmosphere and Ocean
(JISAO) under NOAA Cooperative Agreement #NA67RJO155, Contribution #832, and
#2331 from the NOAA/Pacific Marine Environmental Laboratory. This is U.S. JGOFS
Contribution Number 683.
Science
Team and the JGOFS and WOCE investigators for making their data available for
this work. We thank Lisa Dilling of the National Oceanic and Atmospheric Administration
(NOAA) Office of Global Programs, Don Rice of the National Science Foundation
and Mike Riches of the Department of Energy (DOE) for their efforts in coordinating
this research. This work was supported by DOE and NOAA as a contribution to
the U.S. JGOFS Synthesis and Modeling Project (Grant No. GC99-220) and by grants
to Taro Takahashi from NSF (OPP-9506684) and NOAA (NA16GP01018). This publication
was supported by the Joint Institute for the Study of the Atmosphere and Ocean
(JISAO) under NOAA Cooperative Agreement #NA67RJO155, Contribution #832, and
#2331 from the NOAA/Pacific Marine Environmental Laboratory. This is U.S. JGOFS
Contribution Number 683.
Atlas of Surface Marine Data, 1994: CD-ROM NODC-56, Ocean Climate Laboratory, NOAA.
Bates, N.R., 2001: Interannual variability of oceanic CO![]() and
biogeochemical properties in the Western North Atlantic subtropical gyre. Deep-Sea
Res. II, 48(8–9), 1507–1528.
and
biogeochemical properties in the Western North Atlantic subtropical gyre. Deep-Sea
Res. II, 48(8–9), 1507–1528.
Battle, M., M.L. Bender, P.P. Tans, J.W.C. White, J.T. Ellis, T. Conway and
R.J. Francey, 2000: Global carbon sinks and their variability inferred from
atmospheric O![]() and
and ![]()
![]() C. Science,
287, 2467–2470.
C. Science,
287, 2467–2470.
Brewer, P.G., 1978: Direct observation of oceanic CO![]() increase. Geophys.
Res. Lett., 5, 997–1000.
increase. Geophys.
Res. Lett., 5, 997–1000.
Chen, C.T. and F.J. Millero, 1979: Gradual increase of oceanic CO![]() . Nature,
277, 205–206.
. Nature,
277, 205–206.
Feely, R.A., R. Wanninkhof, T. Takahashi and P. Tans, 1999: The influence
of the equatorial Pacific contribution to atmospheric CO![]() accumulation. Nature,
398, 597–601.
accumulation. Nature,
398, 597–601.
GLOBALVIEW-CO![]() :
Cooperative Atmospheric Data Integration Project—Carbon Dioxide, 2000. CDROM,
NOAA CMDL, Boulder, CO. Also available on Internet via anonymous FTP to ftp.cmdl.noaa.gov,
Path: ccg/co2/GLOBALVIEW.
:
Cooperative Atmospheric Data Integration Project—Carbon Dioxide, 2000. CDROM,
NOAA CMDL, Boulder, CO. Also available on Internet via anonymous FTP to ftp.cmdl.noaa.gov,
Path: ccg/co2/GLOBALVIEW.
Gruber, N., J.L. Sarmiento and T.F. Stocker, 1996: An improved method for
detecting anthropogenic CO![]() in
the oceans. Global Biogeochem. Cycles, 10, 809–837.
in
the oceans. Global Biogeochem. Cycles, 10, 809–837.
Gruber, N., 1998: Anthropogenic CO![]() in
the Atlantic Ocean. Global Biogeochem. Cycles, 12, 165–191.
in
the Atlantic Ocean. Global Biogeochem. Cycles, 12, 165–191.
Inoue, H.Y., H. Mastueda, M. Ishii, K. Fushimi, M. Hirota, I. Asanuma and
Y. Takasugi, 1995: Longterm trend of the partial pressure of carbon dioxide
(pCO![]() )
in surface waters of the western North Pacific 1984–1993. Tellus,
47B, 391–413.
)
in surface waters of the western North Pacific 1984–1993. Tellus,
47B, 391–413.
Keeling, C.D. and T.P. Whorf, 2000: Atmospheric CO![]() concentrations—Mauna
Loa Observatory, Hawaii, http://cdiac.esd.ornl.gov/npds/npd001.html, Carbon
dioxide Information and Analysis Center, Oak Ridge, TN.
concentrations—Mauna
Loa Observatory, Hawaii, http://cdiac.esd.ornl.gov/npds/npd001.html, Carbon
dioxide Information and Analysis Center, Oak Ridge, TN.
Keeling, R., S.C. Piper and M. Heinmann, 1996: Global and hemispheric CO![]() sinks
deduced from changes in atmospheric O
sinks
deduced from changes in atmospheric O![]() concentration. Nature,
381, 218–221.
concentration. Nature,
381, 218–221.
Liss, P.S. and L. Merlivat, 1986: Air-sea gas exchange rates: Introduction and synthesis. In: The Role of Air-Sea Exchange in Geochemical Cycling. P. Buat-Menard, ed., Reidel, Boston, 113–129.
Nightingale, P.D., G. Malin, C.S. Law, A.J. Watson, P.S. Liss, M.I. Liddicoat, J. Boutin and R.C. Upstill-Goddard, 2000: In situ evaluation of air-sea gas exchange parameterizations using novel conservative and volatile tracers. Global Biogeochem. Cycles, 14, 373–387.
Sabine, C.L. and R.A. Feely, 2001: Comparison of recent Indian Ocean anthropogenic
CO![]() estimates
with a historical approach. Global Biogeochem. Cycles, 15(1), 31–42.
estimates
with a historical approach. Global Biogeochem. Cycles, 15(1), 31–42.
Sarmiento, J.L., R. Murnane and C. LeQuere, 1995: Airsea CO![]() transfer
and the carbon budget of the North Atlantic. Philos. Trans. R. Soc. Lond. (B
Biol. Sci.), 348, 211–219.
transfer
and the carbon budget of the North Atlantic. Philos. Trans. R. Soc. Lond. (B
Biol. Sci.), 348, 211–219.
Siegenthaler, U. and J.L. Sarmiento, 1993: Atmospheric carbon dioxide and the ocean. Nature, 365, 119–125.
Takahashi, T., S.C. Sutherland, C. Sweeney, A. Poisson, N. Metzl, B. Tillbrook,
N. Bates, R. Wanninkhof, R.A. Feely, C. Sabine, J. Olafsson and Y. Nojiri,
2002: Global sea-air CO![]() flux
based on climatological surface ocean pCO
flux
based on climatological surface ocean pCO![]() ,
and seasonal biological and temperature effects, Deep-Sea Res. II,
49(9–10),
1601–1623.
,
and seasonal biological and temperature effects, Deep-Sea Res. II,
49(9–10),
1601–1623.
Wanninkhof, R., 1992: Relationship between gas exchange and wind speed over the ocean. J. Geophys. Res., 97, 7373–7381.
Wanninkhof, R. and W.M. McGillis, 1999: A cubic relationship between gas transfer and wind speed. Geophys. Res. Lett., 26, 1889–1893.
Wanninkhof, R., S. Doney, T. Takahashi and W. McGillis, 2001: The Effect of
Using Time-Averaged Winds on Regional Air-Sea CO![]() Fluxes.
In: Gas Transfer at Water Surfaces. M.A. Donelan, W.M. Drennan, E.S.
Saltzman and R. Wanninkhof, eds., AGU, Washington, Geophys. Mono. Ser. Vol.
127.
Fluxes.
In: Gas Transfer at Water Surfaces. M.A. Donelan, W.M. Drennan, E.S.
Saltzman and R. Wanninkhof, eds., AGU, Washington, Geophys. Mono. Ser. Vol.
127.
Return to Abstract